
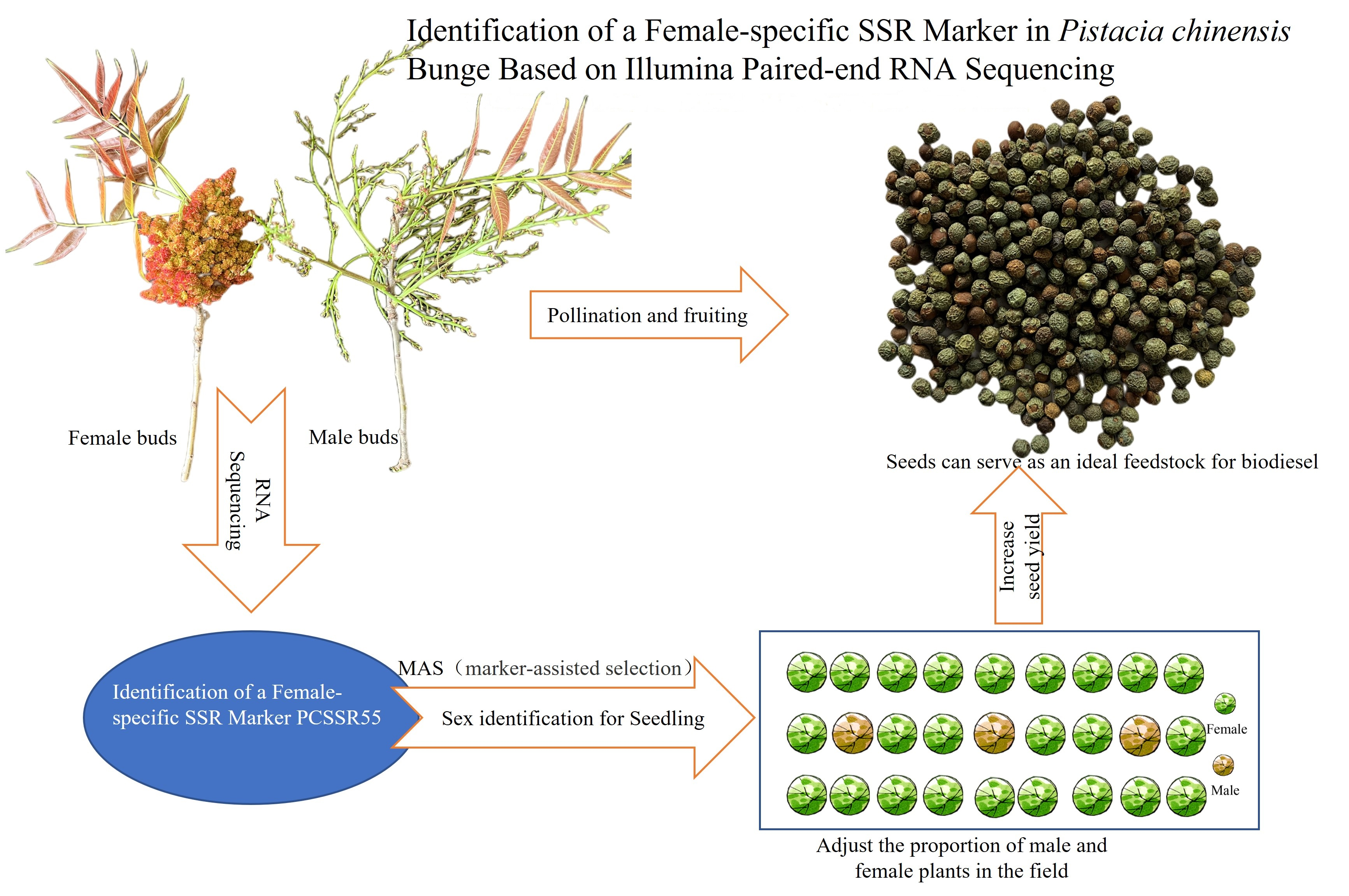
Transcriptome Analysis and Identification of a Female-Specific SSR Marker in Pistacia chinensis Based on Illumina Paired-End RNA Sequencing


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Sampling and RNA Extraction
2.2. cDNA Library Construction and Paired-End Sequencing
2.3. Data Pre-Processing and De Novo Assembly
2.4. Functional Annotation
2.5. Simple Sequence Repeat Discovery and Primer Design
2.6. DNA Extraction and EST-SSR Marker Evaluation
3. Results
3.1. Illumina Paired-End Sequencing and De Novo Assembly of P. chinensis Transcriptome
3.2. Sequence Annotation of P. chinensis Transcriptome
3.3. KEGG Pathway Assignment of Unigenes
3.4. Molecular Characterization of SSR Motifs
3.5. Identification of Sex-Linked SSR Markers
4. Discussion
4.1. Characterization of P. chinensisTranscriptome
4.2. Abundance and Distribution of SSR Motifs
4.3. Validation and Polymorphism of EST-SSRs for Gender Identification of P. chinensis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karmakar, A.; Karmakar, S.; Mukherjee, S. Properties of various plants and animals feed stocks for biodiesel production. Bioresour. Technol. 2010, 101, 7201–7210. [Google Scholar] [CrossRef] [PubMed]
- Kafkas, S.; Özkan, H.; Ak, B.E.; Acar, I.; Atli, H.S.; Koyuncu, S. Detecting DNA polymorphism and genetic diversity in a wide pistachio germplasm: Comparison of AFLP, ISSR and RAPD markers. J. Am. Soc. Hortic. Sci. 2006, 131, 522–529. [Google Scholar] [CrossRef]
- Qin, S.J.; Sun, Y.Z.; Meng, X.C.; Zhang, S.X. Production and analysis of biodiesel from non-edible seed oil of Pistacia chinensis. Energy Explor. Exploit. 2010, 28, 37–46. [Google Scholar] [CrossRef]
- Wang, L.B.; Yu, H.Y.; He, X.H. Assessment on fuel properties of four woody biodiesel plants species in China. Sci. Silvae Sin. 2012, 48, 150–154. [Google Scholar]
- Hormaza, J.I.; Dollo, L.; Polito, V.S. Identification of a RAPD marker linked to sex determination in Pistacia vera using bulked segregant analysis. Theor. Appl. Genet. 1994, 89, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Yakubov, B.; Barazani, O.; Golan-Goldhirsh, A. Combination of SCAR primers and touchdown-PCR for sex identification in Pistacia vera L. Sci. Hortic. 2005, 103, 473–478. [Google Scholar] [CrossRef]
- Esfandiyari, B.; Davarynejad, G.H.; Shahriari, F.; Kiani, M.; Mathe, A. Data to the sex determination in Pistacia species using molecular markers. Euphytica 2012, 185, 227–231. [Google Scholar] [CrossRef]
- Kafkas, S.; Khodaeiaminjan, M.; Güney, M.; Kafkas, E. Identification of sex-linked SNP markers using RAD sequencing suggests ZW/ZZ sex determination in Pistacia vera L. BMC Genom. 2015, 16, 98. [Google Scholar] [CrossRef] [Green Version]
- Khodaeiaminjan, M.; Kafkas, E.; Güney, M.; Kafkas, S. Development and linkage mapping of novel sex-linked markers for marker-assisted cultivar breeding in pistachio (Pistacia vera L.). Mol. Breed. 2017, 37, 98. [Google Scholar] [CrossRef]
- Sun, Q.; Yang, X.; Li, R. SCAR marker for sex identification of Pistacia chinensis Bunge (Anacardiaceae). Genet. Mol. Res. 2014, 13, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Chen, M.; Xu, C.J.; Bai, L.; Yin, X.R.; Li, X.; Allan, A.C.; Ferguson, I.B.; Chen, K.S. Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genom. 2012, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.D.; He, S.P. Transcriptome analysis of silver carp (Hypophthalmichthys molitrix) by paired-end RNA sequencing. DNA Res. 2012, 19, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, A.; Mullick, T.; Schliesky, S.; Weber, A. Critical assessment of assembly strategies for non-model species mRNA-Seq data and application of next-generation sequencing to the comparison of C (3) and C (4) species. J. Exp. Bot. 2011, 62, 3093–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, D.A.; Ragland, G.J.; Shoemaker, D.D.; Denlinger, D.L. Gene discovery using massively parallel pyrosequencing to develop ESTs for the flesh fly Sarcophaga crassipalpis. BMC Genom. 2009, 10, 234. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.X.; He, D.; Dong, W.R.; Zhang, Y.W.; Shao, J.Z. Deep sequencing-based transcriptome profiling analysis of bacteria-challenged Lateolabrax japonicus reveals insight into the immune-relevant genes in marine fish. BMC Genom. 2010, 11, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierocka, I.; Alaba, S.; Jarmolowski, A.; Karlowski, W.M.; Szweykowska-Kulinska, Z. The identification of differentially expressed genes in male and female gametophytes of simple thalloid liverwort Pellia endiviifolia sp. B using an RNA-seq approach. Planta 2020, 252, 21. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.J.N.; Coito, J.; Fino, J.; Cunha, J.; Silva, H.; de Almeida, P.G.; Costa, M.M.R.; Amancio, S.; Paulo, O.S.; Rocheta, M. Deep analysis of wild Vitis flower transcriptome reveals unexplored genome regions associated with sex specification. Plant Mol. Biol. 2017, 93, 151–170. [Google Scholar] [CrossRef] [Green Version]
- Prentout, D.; Razumova, O.; Rhone, B.; Badouin, H.; Henri, H.; Feng, C.; Kafer, J.; Karlov, G.; Marais, G.A.B. An efficient RNA-seq-based segregation analysis identifies the sex chromosomes of Cannabis sativa. Genome Res. 2020, 30, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Sheng, Y.Y.; Luan, F.S.; Ma, H.Y.; Liu, S. RNA-Seq transcriptome profiling reveals differentially expressed genes involved in sex expression in melon. Crop Sci. 2015, 55, 1686–1695. [Google Scholar] [CrossRef]
- Xin, G.L.; Liu, J.Q.; Liu, J.; Ren, X.L.; Du, X.M.; Liu, W.Z. Anatomy and RNA-Seq reveal important gene pathways regulating sex differentiation in a functionally Androdioecious tree, Tapiscia sinensis. BMC Plant Biol. 2019, 19, 554. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; Andre, C.; Hanafey, M.; Vogel, J.; Tingey, S.; Rafalski, A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 1996, 2, 225–238. [Google Scholar] [CrossRef]
- Choi, K.Y.; Park, D.H.; Seong, E.S.; Sang, W.L.; Na, J.K. Transcriptome analysis of a medicinal plant, Pistacia chinensis. J. Plant Biotechnol. 2019, 46, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Pertea, G.; Huang, X.Q.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Fang, L.; Zheng, H.K.; Zhang, Y.; Chen, J.; Zhang, Z.J.; Wang, J.; Li, S.T.; Li, R.Q.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Koressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef]
- Cheng, X.M.; Xu, J.S.; Xia, S.; Gu, J.X.; Yang, Y.; Fu, J.; Qian, X.J.; Zhang, S.C.; Wu, J.S.; Liu, K.D. Development and genetic mapping of microsatellite markers from genome survey sequences in Brassica napus. Theor. Appl. Genet. 2009, 118, 1121–1131. [Google Scholar] [CrossRef]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. In Proceedings of the Seventh International Conference on Intelligent Systems for Molecular Biology, Heidelberg, Germany, 6–10 August 1999; pp. 138–148. [Google Scholar]
- Wei, Z.; Sun, Z.; Cui, B.; Zhang, Q.; Xiong, M.; Wang, X.; Zhou, D. Transcriptome analysis of colored calla lily (Zantedeschia rehmannii Engl.) by Illumina sequencing: De novo assembly, annotation and EST-SSR marker development. PeerJ 2016, 4, e2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Xue, L.; Guo, R.X.; Luo, G.J.; Song, Y.T.; Lei, J.J. De Novo assembled transcriptome analysis and identification of genic SSR markers in red-flowered strawberry. Biochem. Genet. 2019, 57, 607–622. [Google Scholar] [CrossRef]
- Zeng, L.; Tu, X.L.; Dai, H.; Han, F.M.; Lu, B.S.; Wang, M.S.; Nanaei, H.A.; Tajabadipour, A.; Mansouri, M.; Li, X.L.; et al. Whole genomes and transcriptomes reveal adaptation and domestication of pistachio. Genome Biol. 2019, 20, 79. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.P.; Yang, T.; Liu, R.; Hu, J.G.; Yao, Y.; Burlyaeva, M.; Wang, Y.; Ren, G.X.; Zhang, H.Y.; Wang, D.; et al. An RNA sequencing transcriptome analysis of Grasspea (Lathyrus sativus L.) and development of SSR and KASP markers. Front. Plant Sci. 2017, 8, 1873. [Google Scholar] [CrossRef]
- Mei, L.; Dong, N.; Li, F.; Li, N.; Yao, M.; Chen, F.; Tang, L. Transcriptome analysis of female and male flower buds of Idesia polycarpa Maxim. var. vestita Diels. Electron. J. Biotechnol. 2017, 29, 39–46. [Google Scholar] [CrossRef]
- Chen, H.L.; Wang, L.L.; Wang, S.H.; Somta, P.; Cheng, X.Z. Development and Validation of EST-SSR Markers from the Transcriptome of Adzuki Bean (Vigna angularis). PLoS ONE 2015, 10, e0131939. [Google Scholar] [CrossRef] [Green Version]
- Taheri, S.; Abdullah, T.L.; Rafii, M.Y.; Harikrishna, J.A.; Werbrouck, S.P.O.; Teo, C.H.; Sahebi, M.; Azizi, P. De novo assembly of transcriptomes, mining, and development of novel EST-SSR markers in Curcuma alismatifolia (Zingiberaceae family) through Illumina sequencing. Sci. Rep. 2019, 9, 3047. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Wang, Z.; Wang, T.; Su, Y. Characterization and application of EST-SSR markers developed from the transcriptome of Amentotaxus argotaenia (Taxaceae), a relict vulnerable conifer. Front. Genet. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.B.; Liu, Y.L.; Xiong, B.; Jiang, X.N.; Zhang, Z.X. Transcriptomic analysis of a potential bioenergy tree, Pistacia chinensis Bunge, and identification of candidate genes involved in the biosynthesis of oil. Bioenergy Res. 2016, 9, 740–749. [Google Scholar] [CrossRef]
- Moazzzam Jazi, M.; Mahdi Seyedi, S.; Ebrahimie, E.; Ebrahimi, M.; De Moro, G.; Botanga, C. A genome-wide transcriptome map of pistachio (Pistacia vera L.) provides novel insights into salinity-related genes and marker discovery. BMC Genom. 2017, 18, 627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, M.; Hou, L.; Zhang, Z.Y.; Li, Y.Y. De novo transcriptome assembly and population genetic analyses for an endangered chinese endemic Acer miaotaiense (Aceraceae). Genes 2018, 9, 378. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Fillmore, N.; Bai, Y.S.; Collins, M.; Thomson, J.A.; Stewart, R.; Dewey, C.N. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 2014, 15, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzberg, S.L.; Phillippy, A.M.; Zimin, A.; Puiu, D.; Magoc, T.; Koren, S.; Treangen, T.J.; Schatz, M.C.; Delcher, A.L.; Roberts, M.; et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012, 22, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly, annotation, and marker discovery. BMC Genom. 2010, 11, 180. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Li, Z.H.; Bai, G.Q.; Feng, L.; Chen, C.; Wei, Y.; Chang, Y.X.; Zhao, G.F. Transcriptome sequencing and development of genic SSR markers of an endangered Chinese endemic genus Dipteronia oliver (Aceraceae). Molecules 2016, 21, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.X.; Chen, X.X.; Xu, B.; Li, Y.X.; Ma, Y.H.; Wang, G.D. Phenotype and transcriptome analysis reveals chloroplast development and pigment biosynthesis together influenced the leaf color formation in mutants of Anthuriuman draeanum ‘Sonate’. Front. Plant Sci. 2015, 6, 139. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.M.; Lei, Y.; Yan, L.Y.; Wan, L.Y.; Cai, Y.; Yang, Z.F.; Lv, J.W.; Zhang, X.J.; Xu, C.W.; Liao, B.H. Development and validation of simple sequence repeat markers from Arachi shypogaea transcript sequences. Crop. J. 2018, 6, 172–180. [Google Scholar] [CrossRef]
- Al-Qurainy, F.; Alshameri, A.; Gaafar, A.R.; Khan, S.; Nadeem, M.; Alameri, A.A.; Tarroum, M.; Ashraf, M.; Kurabayashi, A. Comprehensive stress-based De Novo transcriptome assembly and annotation of guar (Cyamopsis tetragonoloba (L.) Taub.): An important industrial and forage crop. Int. J. Genom. 2019, 2019, 7295859. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Chen, J.; Kou, Y.X.; Wang, Y.J. Application of EST-SSR markers developed from the transcriptome of Torreya grandis (Taxaceae), a threatened nut-yielding conifer tree. PeerJ 2018, 6, e5606. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, P.; Singh, A.; Sheikh, G.; Mahajan, V.; Gupta, A.P.; Bedi, Y.S.; Gandhi, S.G. Mining and characterization of EST-SSR markers for Zingiber officinale Roscoe with transferability to other species of Zingiberaceae. Physiol. Mol. Biol. Plants 2017, 23, 925–931. [Google Scholar] [CrossRef]
- Kumpatla, S.P.; Mukhopadhyay, S. Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome 2005, 48, 985–998. [Google Scholar] [CrossRef]
- Zheng, X.F.; Pan, C.; Diao, Y.; You, Y.N.; Yang, C.Z.; Hu, Z.L. Development of microsatellite markers by transcriptome sequencing in two species of Amorphophallus (Araceae). BMC Genom. 2013, 14, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liu, C.Y.; Liu, Y.F.; VanBuren, R.; Zhong, C.; Huang, H. High-density interspecific genetic maps of kiwifruit and the identification of sex-specific markers. DNA Res. 2015, 22, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammad, M.; Jaskani, M.J.; Awan, F.S.; Ahmad, S.; Khan, I.A. Development of molecular method for sex identification in date palm (Phoenix dactylifera L.) plantlets using novel sex-linked microsatellite markers. 3 Biotech 2016, 6, 22. [Google Scholar]
- Jia, H.M.; Jiao, Y.; Wang, G.Y.; Li, Y.H.; Jia, H.J.; Wu, H.X.; Chai, C.Y.; Dong, X.; Guo, Y.P.; Zhang, L.P.; et al. Genetic diversity of male and female Chinese bayberry (Myrica rubra) populations and identification of sex-associated markers. BMC Genom. 2015, 16, 394. [Google Scholar] [CrossRef] [Green Version]
- Cherif, E.; Zehdi, S.; Castillo, K.; Chabrillange, N.; Abdoulkader, S.; Pintaud, J.C.; Santoni, S.; Salhi-Hannachi, A.; Glémin, S.; Aberlenc-Bertossi, F. Male-specific DNA markers provide genetic evidence of an XY chromosome system, a recombination arrest and allow the tracing of paternal lineages in date palm. New Phytol. 2013, 197, 409–415. [Google Scholar] [CrossRef]
- Zhou, X.J.; Wang, Y.Y.; Xu, Y.N.; Yan, R.S.; Zhao, P.; Liu, W.Z. De novo characterization of flower bud transcriptomes and the development of EST-SSR markers for the endangered tree Tapiscia sinensis. Int. J. Mol. Sci. 2015, 16, 12855–12870. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Total Raw Reads | Total Clean Reads | Total Clean Nucleotides (nt) | Q20 | Q30 | GC Content |
|---|---|---|---|---|---|---|
| PC | 54,895,796 | 50,925,088 | 7638,763,200 | 98.29% | 96.56% | 43.52% |
| PX | 55,210,442 | 51,470,578 | 7720,586,700 | 98.25% | 96.43% | 43.43% |
| Item | Total Number (nt) | Total Length (nt) | Mean Length (nt) | N50 | Distinct Clusters | Distinct Singletons |
|---|---|---|---|---|---|---|
| PC_Contigs | 89,442 | 93,938,149 | 1050 | 1739 | ||
| PX_Contigs | 89,724 | 96,205,186 | 1072 | 1768 | ||
| PC_Unigenes | 65,520 | 80,247,826 | 1225 | 1903 | 29,689 | 35,831 |
| PX_Unigenes | 65,752 | 82,513,165 | 1255 | 1933 | 30,903 | 34,849 |
| All_Unigenes | 83,370 | 110,503,948 | 1325 | 2027 | 42,960 | 40,410 |
| Sequence File | NR | NT | Swiss-Prot | KEGG | COG | GO | All Annotated Unigenes | All Assembled Unigenes |
|---|---|---|---|---|---|---|---|---|
| Number of Unigenes (singleton, cluster, unigene) | 58,543 (20,307, 38,236) | 59,316 (21,071, 38,245) | 38,879 (13,566, 25,313) | 36,136 (12,125, 24,011) | 47,049 (13,999, 33,050) | 40,643 (12,965, 27,678) | 64,539 (24,634, 39,905) | 83,370 (40,410, 42,960) |
| Annotated/All-Unigene (%) | 70.22 | 71.15 | 46.63 | 43.34 | 56.43 | 48.75 | 77.48 |
| Rank | Pathway | Genes within the Coverage of Pathway Annotations (n = 36,136) | Pathway ID |
|---|---|---|---|
| 1 | Metabolic pathways | 8208 (22.71%) | ko01100 |
| 2 | Biosynthesis of secondary metabolites | 4045 (11.19%) | ko01110 |
| 3 | Plant-pathogen interaction | 2516 (6.96%) | ko04626 |
| 4 | Plant hormone signal transduction | 1729 (4.78%) | ko04075 |
| 5 | Spliceosome | 1336 (3.7%) | ko03040 |
| 6 | RNA transport | 1203 (3.33%) | ko03013 |
| 7 | Pyrimidine metabolism | 1096 (3.03%) | ko00240 |
| 8 | Purine metabolism | 1086 (3.01%) | ko00230 |
| 9 | Ribosome | 981 (2.71%) | ko03010 |
| 10 | Protein processing in endoplasmic reticulum | 962 (2.66%) | ko04141 |
| 11 | Endocytosis | 811 (2.24%) | ko04144 |
| 12 | Starch and sucrose metabolism | 757 (2.09%) | ko00500 |
| 13 | Ubiquitin mediated proteolysis | 746 (2.06%) | ko04120 |
| 14 | RNA polymerase | 719 (1.99%) | ko03020 |
| 15 | Ribosome biogenesis in eukaryotes | 684 (1.89%) | ko03008 |
| 16 | Glycerophospholipid metabolism | 677 (1.87%) | ko00564 |
| 17 | RNA degradation | 648 (1.79%) | ko03018 |
| 18 | mRNA surveillance pathway | 600 (1.66%) | ko03015 |
| 19 | Phenylpropanoid biosynthesis | 574 (1.59%) | ko00940 |
| 20 | Glycolysis/Gluconeogenesis | 530 (1.47%) | ko00010 |
| Searching Item | Numbers |
|---|---|
| Total number of examined sequences | 83,370 |
| Total size of examined sequences (bp) | 110,503,948 |
| Total number of identified SSR markers | 21,662 |
| Number of SSR-containing sequences | 17,028 |
| Number of sequences with >1 SSR | 3545 |
| Number of SSR markers found in compound formation | 1376 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, X.; Wang, F.; Luo, W.; Kuang, J.; Huang, X. Transcriptome Analysis and Identification of a Female-Specific SSR Marker in Pistacia chinensis Based on Illumina Paired-End RNA Sequencing. Genes 2022, 13, 1024. https://doi.org/10.3390/genes13061024
Cheng X, Wang F, Luo W, Kuang J, Huang X. Transcriptome Analysis and Identification of a Female-Specific SSR Marker in Pistacia chinensis Based on Illumina Paired-End RNA Sequencing. Genes. 2022; 13(6):1024. https://doi.org/10.3390/genes13061024
Chicago/Turabian StyleCheng, Xiaomao, Fei Wang, Wen Luo, Jingge Kuang, and Xiaoxia Huang. 2022. "Transcriptome Analysis and Identification of a Female-Specific SSR Marker in Pistacia chinensis Based on Illumina Paired-End RNA Sequencing" Genes 13, no. 6: 1024. https://doi.org/10.3390/genes13061024