
Molecular Diversity and Phylogeny Reconstruction of Genus Colobanthus (Caryophyllaceae) Based on Mitochondrial Gene Sequences

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, DNA Extraction and Mitochondrial Genome Sequencing
2.2. Gene Annotation and Comparative Analysis
2.3. Phylogenetic Analysis
3. Results
3.1. Mitochondrial Genes Assembly, Annotation and Characteristics
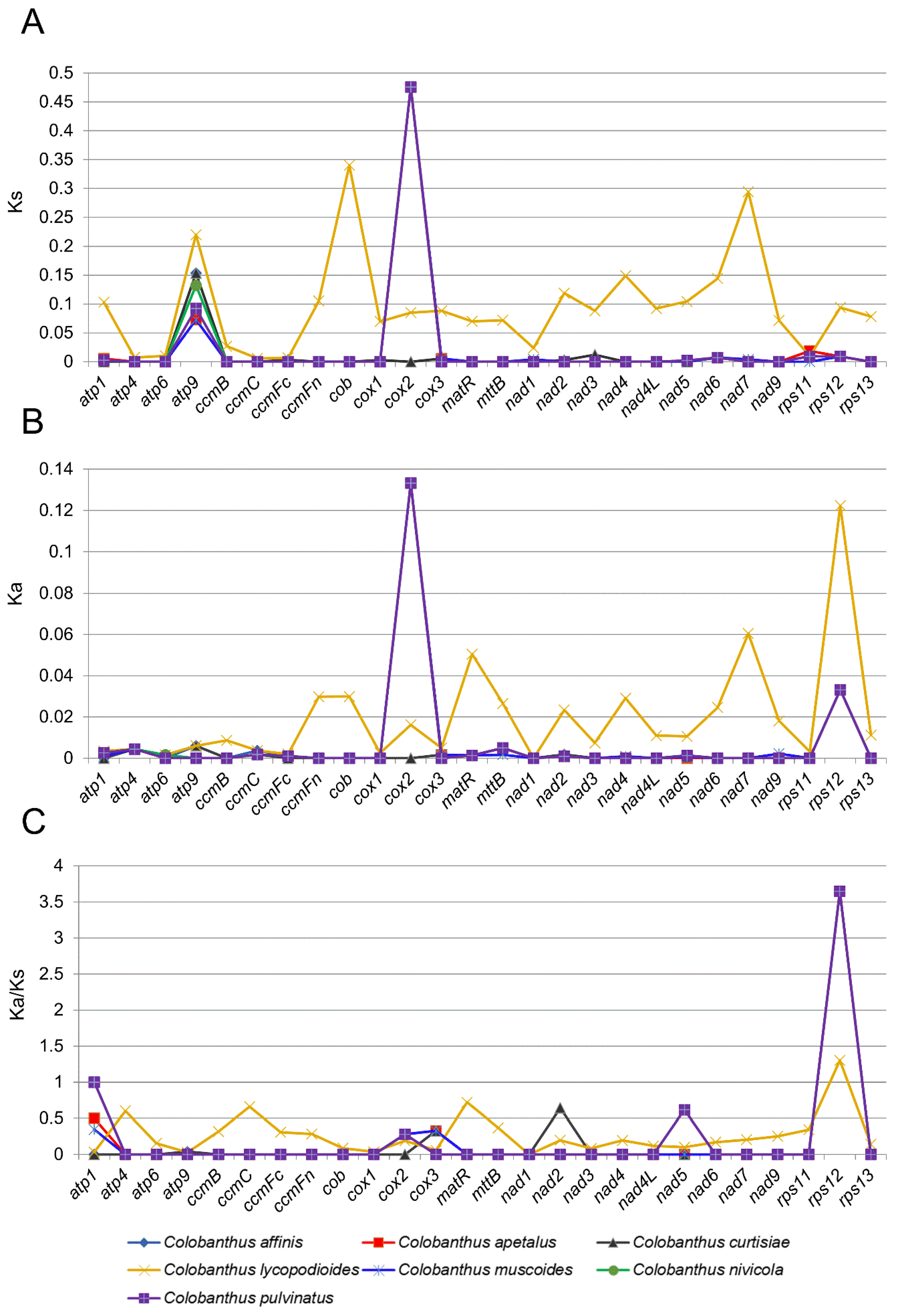
3.2. Synonymous (Ks) and Non-Synonymous (Ka) Substitution Rate Analysis
3.3. RNA Editing Sites
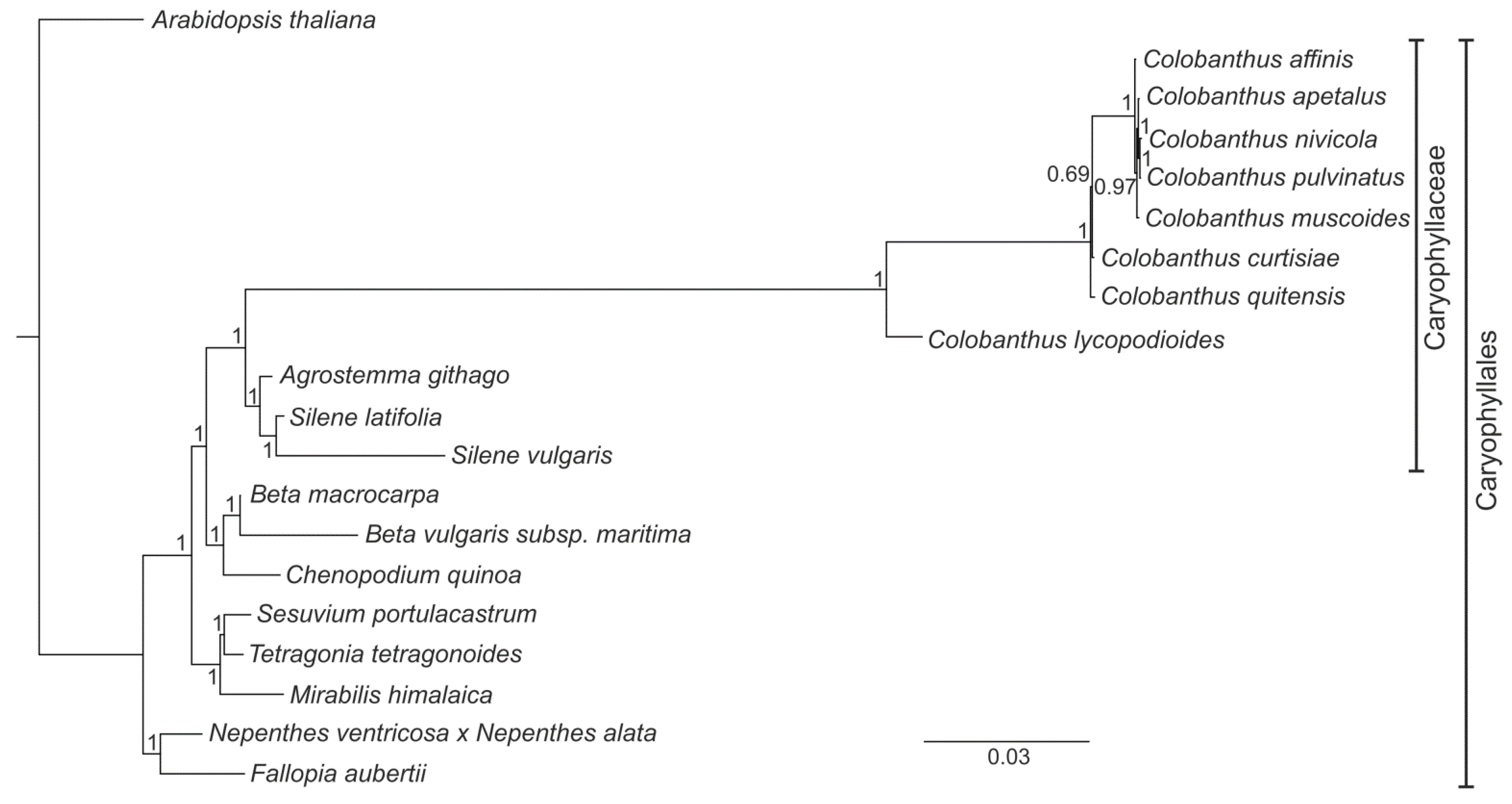
3.4. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christenhusz, M.J.M.; Byng, J.W. The number of known plants species in the world and its annual increase. Phytotaxa 2016, 261, 201–217. [Google Scholar] [CrossRef] [Green Version]
- The Plant List. Version 1.1. Available online: http://www.theplantlist.org/browse/A/Caryophyllaceae/Colobanthus (accessed on 3 February 2022).
- West, J.G. Colobanthus curtisiae (Caryophyllaceae), a new species from eastern Tasmania, Australia. Pap. Proc. R. Soc. Tasman. 1991, 124, 75–78. [Google Scholar] [CrossRef]
- The AGS Online Plant Encyclopaedia. Alpine Garden Society. Available online: http://encyclopaedia.alpinegardensociety.net/plants/Colobanthus (accessed on 3 February 2022).
- Harbaugh, D.T.; Nepokroeff, M.; Rabeler, R.K.; McNeill, J.; Zimmer, E.A.; Wagner, W.L. A new lineage-based tribal classification of the family Caryophyllaceae. Int. J. Plant Sci. 2010, 171, 185–198. [Google Scholar] [CrossRef] [Green Version]
- Greene, S.W.; Greene, D.M. Check list of the sub-Antarctic and Antarctic vascular flora. Polar Rec. 1963, 11, 411–418. [Google Scholar] [CrossRef]
- Moore, D.M. Studies in Colobanthus quitensis (Kunth) Bartl. and Deschampsia antarctica Desv.: II. Taxonomy, distribution and relationships. Br. Antarct. Surv. B. 1970, 23, 63–80. [Google Scholar]
- Parnikoza, I.; Kozeretska, I.; Kunakh, V. Vascular plants of the maritime Antarctic: Origin and adaptation. Am. J. Plant Sci. 2011, 2, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Briggs, J.D.; Leigh, J.H. Rare or Threatened Australian Plants; CSIRO Publishing: Collingwood, Australia, 1996. [Google Scholar]
- Gray, M. Miscellaneous notes on Australian plants. 3. Craspedia, Gnaphalium, Epacris, Tasmannia, Colobanthus and Deyeuxia. Contr. Herb. Aust. 1976, 1976, 1–11. [Google Scholar]
- Gilfedder, L.; Kirkpatrick, J.B. The distribution, ecology and conservation needs of Colobanthus curtisiae west. Pap. Proc. R. Soc. Tasman. 1996, 130, 25–30. [Google Scholar] [CrossRef]
- Sneddon, B.V. The taxonomy and breeding system of Colobanthus squarrosus (Caryophyllaceae). N. Z. J. Bot. 1999, 37, 195–204. [Google Scholar] [CrossRef]
- Gianoli, E.; Inostroza, P.; Zuniga-Feest, A.; Reyes-Diaz, M.; Cavieres, L.A.; Bravo, L.A.; Corcuera, L.J. Ecotypic differentiation in morphology and cold resistance in populations of Colobanthus quitensis (Caryophyllaceae) from the Andes of central Chile and the Maritime Antarctic. Arct. Antarct. Alp. Res. 2004, 36, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, A.D.; Hardwick, K. Evolution and stress-genotypic and phenotypic components. Biol. J. Linn. Soc. 1989, 37, 137–155. [Google Scholar] [CrossRef]
- Ng, S.; De Clercq, I.; Van Aken, O.; Law, S.R.; Ivanova, A.; Willems, P.; Giraud, E.; Van Breusegem, F.; Whelan, J. Anterograde and retrograde regulation of nuclear genes encoding mitochondrial proteins during growth, development, and stress. Mol. Plant 2014, 7, 1075–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant. Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef] [Green Version]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef] [Green Version]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef] [Green Version]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 654. [Google Scholar] [CrossRef]
- Laroche, J.; Li, P.; Maggia, L.; Bousquet, J. Molecular evolution of angiosperm mitochondrial introns and exons. Proc. Natl. Acad. Sci. USA 1997, 94, 5722–5727. [Google Scholar] [CrossRef] [Green Version]
- Satoh, M.; Kubo, T.; Nishizawa, S.; Estiati, A.; Itchoda, N.; Mikami, T. The cytoplasmic male sterile type and normal type mitochondrial genomes of sugar beet share the same complement of genes of known function but differ in the content of expressed ORFs. Mol. Genet. Genom. 2004, 272, 247–256. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; Mccauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef] [Green Version]
- Kitazaki, K.; Kubo, T. Cost of having the largest mitochondrial genome: Evolutionary mechanism of plant mitochondrial genome. J. Bot. 2010, 2010, 620137. [Google Scholar] [CrossRef] [Green Version]
- Galtier, N. The intriguing evolutionary dynamics of plant mitochondrial DNA. BMC Biol. 2011, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, J. Promiscuous DNA–chloroplast genes inside plant mitochondria. Nature 1982, 299, 678–679. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Newton, K.J. Angiosperm mitochondrial genomes and mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar] [CrossRef]
- Richardson, A.O.; Palmer, J.D. Horizontal gene transfer in plants. J. Exp. Bot. 2007, 58, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bock, R. The give-and-take of DNA: Horizontal gene transfer in plants. Trends Plant Sci. 2010, 15, 11–22. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef]
- Skuza, L.; Filip, E.; Szućko, I. Use of Organelle Markers to Study Genetic Diversity in Soybean. In A Comprehensive Survey of International Soybean Research—Genetics, Physiology, Agronomy and Nitrogen Relationships; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Wang, P.; Cheng, Q.; Sun, L.; Wang, H.; Wang, Y.; Kao, L.; Li, Y.; Qiu, T.; Yang, W.; et al. Proteomic analysis reveals strong mitochondrial involvement in cytoplasmic male sterility of pepper (Capsicum annuum L.). J. Proteom. 2017, 168, 15–27. [Google Scholar] [CrossRef]
- Touzet, P.; Meyer, E.H. Cytoplasmic male sterility and mitochondrial metabolism in plants. Mitochondrion 2014, 19, 166–171. [Google Scholar] [CrossRef]
- Knoop, V. The mitochondrial DNA of land plants: Peculiarities in phylogenetic perspective. Curr. Genet. 2004, 46, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Lee, H.; Kim, M.K.; Shin, S.C.; Park, H.; Lee, J. The complete chloroplast genome of Antarctic pearlwort, Colobanthus quitensis (Kunth) Bartl. (Caryophyllaceae). Mitochondrial DNA Part A 2016, 27, 4677–4678. [Google Scholar] [CrossRef] [PubMed]
- Androsiuk, P.; Jastrzębski, J.P.; Paukszto, Ł.; Okorski, A.; Pszczółkowska, A.; Chwedorzewska, K.J.; Koc, J.; Górecki, R.; Giełwanowska, I. The complete chloroplast genome of Colobanthus apetalus (Labill.) Druce: Genome organization and comparison with related species. PeerJ 2018, 23, e4723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Androsiuk, P.; Jastrzębski, J.P.; Paukszto, Ł.; Makowczenko, K.; Okorski, A.; Pszczółkowska, A.; Chwedorzewska, K.J.; Górecki, R.; Giełwanowska, I. Evolutionary dynamics of the chloroplast genome sequences of six Colobanthus species. Sci. Rep. 2020, 10, 11522. [Google Scholar] [CrossRef]
- Cho, S.M.; Lee, H.; Jo, H.; Lee, H.; Kang, Y.; Park, H.; Lee, J. Comparative transcriptome analysis of field- and chamber-grown samples of Colobanthus quitensis (Kunth) Bartl, an Antarctic flowering plant. Sci. Rep. 2018, 8, 11049. [Google Scholar] [CrossRef] [Green Version]
- Nibert, M.L.; Manny, A.R.; Debat, H.J.; Firth, A.E.; Bertini, L.; Caruso, C. A barnavirus sequence mined from a transcriptome of the Antarctic pearlwort Colobanthus quitensis. Arch. Virol. 2018, 163, 1921–1926. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, G.I.; Torres-Díaz, C.; Bravo, L.A.; Balboa, K.; Caruso, C.; Bertini, L.; Proietti, S.; Molina-Montenegro, M.A. In silico analysis of metatranscriptomic data from the Antarctic vascular plant Colobanthus quitensis: Responses to a global warming scenario through changes in fungal gene expression levels. Fungal Ecol. 2020, 43, 100873. [Google Scholar] [CrossRef]
- West, J.G.; Cowley, K.J. Colobanthus. In Flora of Victoria. Vol. 3, Dicotyledons Winteraceae to Myrtaceae; Walsh, N.G., Entwisle, T.J., Eds.; Inkata Press: Melbourne, Australia, 1996. [Google Scholar]
- Mantowani, A.; Vieira, R.C. Leaf micromorphology of Antarctic pearlwort Colobanthus quitensis (Kunth) Bartl. Polar Biol. 2000, 23, 531–538. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 15, 1757–1764. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Del Barrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef] [PubMed]
- Edera, A.A.; Small, I.; Milone, D.H.; Sanchez-Puerta, M.V. Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 2021, 136, 104682. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P.; Sloan, D.B.; Alverson, A.J. Plant mitochondrial genome diversity: The genomics revolution. In Plant Genome Diversity; Wendel, J.F., Greilhuber, J., Dolezel, J., Leitch, I.J., Eds.; Springer: Vienna, Austria, 2012; pp. 123–144. [Google Scholar]
- Richardson, O.A.; Rice, D.W.; Young, G.J.; Alverson, A.J.; Palmer, J.D. The “fossilized” mitochondrial genome of Liriodendron tulipifera: Ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 2013, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P. Variation in protein gene and intron content among land plant mitogenomes. Mitochondrion 2020, 53, 203–213. [Google Scholar] [CrossRef]
- Adams, K.L.; Qiu, Y.-L.; Stoutemyer, M.; Palmer, J.D. Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. USA 2002, 99, 9905–9912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoop, V. Seed plant mitochondrial genomes: Complexity evolving. In Genomics of Chloroplasts and Mitochondria; Bock, R., Knoop, V., Eds.; Springer: Dordrecht, The Netherland, 2012; pp. 175–200. [Google Scholar]
- Robison, M.M.; Wolyn, D.J. A mitochondrial plasmid and plasmid-like RNA and DNA polymerases encoded within the mitochondrial genome of carrot (Daucus carota L.). Curr. Genet. 2005, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Makalowski, W.; Boguski, M.S. Evolutionary parameters of the transcribed mammalian genome: An analysis of 2820 orthologous rodent and human sequences. Proc. Natl. Acad. Sci. USA 1998, 95, 9407–9412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mower, J.P.; Touzet, P.; Gummow, J.S.; Delph, L.F.; Palmer, J.D. Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol. Biol. 2007, 7, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.B.; Oxelman, B.; Rautenberg, A.; Taylor, D.R. Phylogenetic analysis of mitochondrial substitution rate variation in the angiosperm tribe Sileneae (Caryophyllaceae). BMC Evol. Biol. 2009, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Ruhlman, T.A.; Weng, M.L.; Hajrah, N.H.; Sabir, J.S.M.; Jansen, R.K. Contrasting patterns of nucleotide substitution rates provide insight into dynamic evolution of plastid and mitochondrial genomes of Geranium. Genome Biol. Evol. 2017, 9, 1766–1780. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.H.; Yang, F. Male sterility induction and evolution of cytoplasmic male sterility related atp9 gene from Boehmeria nivea (L.) Gaudich. Ind. Crops Prod. 2020, 156, 112861. [Google Scholar] [CrossRef]
- Park, S.; Grewe, F.; Zhu, A.; Ruhlman, T.A.; Sabir, J.; Mower, J.P.; Jansen, R.K. Dynamic evolution of Geranium mitochondrial genomes through multiple horizontal and intracellular gene transfers. New Phytol. 2015, 208, 570–583. [Google Scholar] [CrossRef]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983. [Google Scholar]
- Cho, Y.; Mower, J.P.; Qiu, Y.L.; Palmer, J.D. Mitochondrial substitution rates are extraordinarily elevated and variable in a genus of flowering plants. Proc. Natl. Acad. Sci. USA 2004, 101, 17741–17746. [Google Scholar] [CrossRef] [Green Version]
- Chamary, J.V.; Parmley, J.L.; Hurst, L.D. Hearing silence: Non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006, 7, 98–108. [Google Scholar] [CrossRef]
- Bazin, E.; Glémin, S.; Galtier, N. Population size does not influence mitochondrial genetic diversity in animals. Science 2006, 312, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, X.; Priyadarshani, S.V.G.N.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.; Zhou, Q.; Luo, Z.; Deng, F.; et al. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Duan, Z.; Wang, Y.; Zhang, Q.; Li, W. Sequence analysis of the aomplete mitochondrial genome of a medicinal plant, Vitex rotundifolia Linnaeus f. (Lamiales: Lamiaceae). Genes 2022, 13, 839. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Zhuang, Y.; Zhang, P.; Adams, K.L. Comparative analysis of structural diversity and sequence evolution in plant mitochondrial genes transferred to the nucleus. Mol. Biol. Evol. 2009, 26, 875–891. [Google Scholar] [CrossRef] [Green Version]
- Giegé, P.; Brennicke, A. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc. Natl. Acad. Sci. USA 1999, 96, 15324–15329. [Google Scholar] [CrossRef] [Green Version]
- Hiesel, R.; Wissinger, B.; Schuster, W.; Brennicke, A. RNA editing in plant mitochondria. Science 1989, 246, 1632–1634. [Google Scholar] [CrossRef]
- Sloan, D.B.; MacQueen, A.H.; Alverson, A.J.; Palmer, J.D.; Taylor, D.R. Extensive loss of RNA editing sites in rapidly evolving Silene mitochondrial genomes: Selection vs. retroprocessing as the driving force. Genetics 2010, 185, 1369–1380. [Google Scholar] [CrossRef] [Green Version]
- Freyer, R.; Kiefer-Meyer, M.C.; Kössel, H. Occurrence of plastid RNA editing in all major lineages of land plants. Proc. Natl. Acad. Sci. USA 1997, 94, 6285–6290. [Google Scholar] [CrossRef] [Green Version]
- Farajollahi, S.; Maas, S. Molecular diversity through RNA editing: A balancing act. Trends Genet. 2010, 26, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Burt, A.; Trivers, R. Genes in Conflict: The Biology of Selfish Genetic Elements; Belknap Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Borner, G.V.; Yokobori, S.; Morl, M.; Dorner, M.; Paabo, S. RNA editing in metazoan mitochondria: Staying fit without sex. FEBS Lett. 1997, 409, 320–324. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M. The Origins of Genome Architecture; Sinauer Associates: Sunderland, MA, USA, 2007. [Google Scholar]
- Soltis, D.E.; Soltis, P.S.; Chase, M.W.; Mort, M.E.; Albach, D.C.; Zanis, M.; Savolainen, V.; Hahn, W.H.; Hoot, S.B.; Fay, M.F.; et al. Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot. J. Linn. Soc. 2000, 133, 381–461. [Google Scholar] [CrossRef]
- Graham, S.W.; Olmstead, R.G. Utility of 17 chloroplast genes for inferring the phylogeny of the basal angiosperms. Am. J. Bot. 2000, 87, 1712–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilu, K.W.; Borsch, T.; Muller, K.; Soltis, D.E.; Soltis, P.S.; Savolainen, V.; Chase, M.W.; Powell, M.P.; Alice, L.A.; Evans, R.; et al. Angiosperm phylogeny based on matK sequence information. Am. J. Bot. 2003, 90, 1758–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, G.; Seberg, O.; Davis, J.I.; Stevenson, D.W. RNA editing and phylogenetic reconstruction in two monocot mitochondrial genes. Taxon 2006, 55, 871–886. [Google Scholar] [CrossRef]
- Zhu, X.Y.; Chase, M.W.; Qiu, Y.L.; Kong, H.Z.; Dilcher, D.L.; Li, J.H.; Chen, Z.D. Mitochondrial matR sequences help to resolve deep phylogenetic relationships in rosids. BMC Evol. Biol. 2007, 7, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Li, L.; Wang, B.; Xue, J.; Hendry, T.A.; Li, R.; Brown, J.W.; Liu, Y.; Hudson, G.T.; Chen, Z. Angiosperm phylogeny inferred from sequences of four mitochondrial genes. J. Syst. Evol. 2010, 48, 391–425. [Google Scholar] [CrossRef]
- Alban, D.M.; Biersma, E.M.; Kadereit, J.W.; Dillenberger, M.S. Colonization of the Southern Hemisphere by Sagina and Colobanthus (Caryophyllaceae). Plant Syst. Evol. 2022, 308, 1. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Accession Number | Species |
|---|---|
| NC_008285 | Arabidopsis thaliana |
| NC_057604 | Agrostemma githago |
| NC_015994 | Beta macrocarpa |
| NC_015099 | Beta vulgaris subsp. maritima |
| NC_041093 | Chenopodium quinoa |
| MW664926 | Fallopia aubertii |
| NC_048974 | Mirabilis himalaica |
| NC_039531 | Nepenthes ventricosa × Nepenthes alata |
| MN683736 | Sesuvium portulacastrum |
| NC_014487 | Silene latifolia |
| JF750427 | Silene vulgaris |
| MW971440 | Tetragonia tetragonoides |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Androsiuk, P.; Paukszto, Ł.; Jastrzębski, J.P.; Milarska, S.E.; Okorski, A.; Pszczółkowska, A. Molecular Diversity and Phylogeny Reconstruction of Genus Colobanthus (Caryophyllaceae) Based on Mitochondrial Gene Sequences. Genes 2022, 13, 1060. https://doi.org/10.3390/genes13061060
Androsiuk P, Paukszto Ł, Jastrzębski JP, Milarska SE, Okorski A, Pszczółkowska A. Molecular Diversity and Phylogeny Reconstruction of Genus Colobanthus (Caryophyllaceae) Based on Mitochondrial Gene Sequences. Genes. 2022; 13(6):1060. https://doi.org/10.3390/genes13061060
Chicago/Turabian StyleAndrosiuk, Piotr, Łukasz Paukszto, Jan Paweł Jastrzębski, Sylwia Eryka Milarska, Adam Okorski, and Agnieszka Pszczółkowska. 2022. "Molecular Diversity and Phylogeny Reconstruction of Genus Colobanthus (Caryophyllaceae) Based on Mitochondrial Gene Sequences" Genes 13, no. 6: 1060. https://doi.org/10.3390/genes13061060
APA StyleAndrosiuk, P., Paukszto, Ł., Jastrzębski, J. P., Milarska, S. E., Okorski, A., & Pszczółkowska, A. (2022). Molecular Diversity and Phylogeny Reconstruction of Genus Colobanthus (Caryophyllaceae) Based on Mitochondrial Gene Sequences. Genes, 13(6), 1060. https://doi.org/10.3390/genes13061060