
Exploring the Potential of Symmetric Exon Deletion to Treat Non-Ischemic Dilated Cardiomyopathy by Removing Frameshift Mutations in TTN


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction/Background
2. The Hypothesis
3. The Titin Gene Contains a Large Number of Potential Target Exons
4. Evaluation of the Hypothesis
5. Consequences of the Hypothesis and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nguyen, Q.; Lim, K.R.Q.; Yokota, T. Genome editing for the understanding and treatment of inherited cardiomyopathies. Int. J. Mol. Sci. 2020, 21, 733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, C.; Fanton, Z.; Tome, M.; Behr, E.R. Inherited cardiomyopathies. BMJ 2019, 365, l1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.G.; Granzier, H.; Mestroni, L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weintraub, R.G.; Christopher Semsarian, P.M. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Braunwald, E. Cardiomyopathies: An overview. Circ. Res. 2017, 121, 711–721. [Google Scholar] [CrossRef]
- Tharp, C.A.; Haywood, M.E.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. The Giant Protein Titin’s Role in Cardiomyopathy: Genetic, Transcriptional, and Post-translational Modifications of TTN and Their Contribution to Cardiac Disease. Front. Physiol. 2019, 10, 1436. [Google Scholar] [CrossRef] [Green Version]
- Azad, A.; Poloni, G.; Sontayananon, N.; Jiang, H.; Gehmlich, K. The giant titin: How to evaluate its role in cardiomyopathies. J. Muscle Res. Cell Motil. 2019, 40, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Gramlich, M.; Pane, L.S.; Zhou, Q.; Chen, Z.; Murgia, M.; Schötterl, S.; Goedel, A.; Metzger, K.; Brade, T.; Parrotta, E.; et al. Antisense-mediated exon skipping: A therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol. Med. 2015, 7, 562–576. [Google Scholar] [CrossRef]
- Herman, D.; Lam, L.; Taylor, M.R.G.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.; McDonough, B.; Sparks, E.; et al. Truncations of Titin Causing Dilated Cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Zou, J.; Tran, D.; Baalbaki, M.; Tang, L.F.; Poon, A.; Pelonero, A.; Titus, E.W.; Yuan, C.; Shi, C.; Patchava, S.; et al. An internal promoter underlies the difference in disease severity between N- and C-terminal truncation mutations of Titin in zebrafish. eLife 2015, 4, e09406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Craig, C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Calvin, C.; et al. Titin Mutations in iPS cells Define Sarcomere Insufficiency as a Cause of Dilated Cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, W.A. Titin Gene and Protein Functions in Passive and Active Muscle. Annu. Rev. Physiol. 2018, 80, 389–411. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Meiler, S.E.; Zhong, T.P.; Mohideen, M.; Crossley, D.A.; Warren, W.; Fishman, M.C. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat. Genet. 2002, 30, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Tabish, A.M.; Knöll, R.; Azzimato, V.; Alexiadis, A.; Buyandelger, B.; Knöll, R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys. Rev. 2017, 9, 207–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiirst, D.O.; Osborn, M.; Nave, R.; Weber, K. The Organization of Titin Filaments in the Half-Sarcomere Revealed by Monoclonal Antibodies in Immunoelectron Microscopy: A Map ofTen Nonrepetitive Epitopes Starting at the Z Line Extends Close to the M Line. J. Cell Biol. 1988, 106, 1563–1572. [Google Scholar] [CrossRef] [Green Version]
- Greaser, M.L.; Guo, W.; Bharmal, S.J.; Esbona, K. Titin diversity alternative splicing gone wild. BioMed Res. Int. 2010, 2010, 753675. [Google Scholar] [CrossRef] [Green Version]
- Yoskovitz, G.; Peled, Y.; Gramlich, M.; Lahat, H. A Novel Titin Mutation in Adult-Onset Familial Dilated Cardiomyopathy. Am. J. Cardiol. 2012, 109, 1644–1650. [Google Scholar] [CrossRef]
- Akinrinade, O.; Koskenvuo, J.W.; Alastalo, T.P. Prevalence of titin truncating variants in general population. PLoS ONE 2015, 10, e0145284. [Google Scholar] [CrossRef] [Green Version]
- Deo, R.C. Alternative Splicing, Internal Promoter, Nonsense-Mediated Decay, or All Three Explaining. Circ. Cardiovasc. Genet. 2016, 9, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Shih, Y.-H.; Dvornikov, A.V.; Zhu, P.; Ma, X.; Kim, M.; Ding, Y.; Xu, X. Exon- and contraction-dependent functions of titin in sarcomere assembly. Development 2016, 143, 4713–4722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Doudna, J.A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016, 34, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.; Lander, E.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Lattanzi, A.; Duguez, S.; Moiani, A.; Izmiryan, A.; Barbon, E.; Martin, S.; Mamchaoui, K.; Mouly, V.; Bernardi, F.; Mavilio, F.; et al. Correction of the Exon 2 Duplication in DMD Myoblasts by a Single CRISPR/Cas9 System. Mol. Ther. Nucleic Acids 2017, 7, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Ceasar, S.A.; Rajan, V.; Prykhozhij, S.V.; Berman, J.N.; Ignacimuthu, S. Insert, remove or replace: A highly advanced genome editing system using CRISPR/Cas9. BBA Mol. Cell Res. 2016, 1863, 2333–2344. [Google Scholar] [CrossRef]
- Lau, C.; Suh, Y. In vivo genome editing in animals using AAV-CRISPR system: Applications to translational research of human disease. F1000Research 2017, 6, 2153. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Chen, M.; Lim, W.A.; Zhao, D.; Qi, L.S. CRISPR/Cas9 for Human Genome Engineering and Disease Research. Annu. Rev. Genom. Hum. Genet. 2016, 17, 131–154. [Google Scholar] [CrossRef] [Green Version]
- Carroll, K.J.; Makarewich, C.A.; Mcanally, J.; Anderson, D.M.; Zentilin, L.; Liu, N.; Giacca, M.; Bassel-Duby, R.; Olson, E.N. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc. Natl. Acad. Sci. USA 2016, 113, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Slaymaker, I.; Gao, L.; Zetsche, B.; Scott, D.; Yan, W.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2015, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Romano, R.; Ghahremani, S.; Zimmerman, T.; Legere, N.; Thakar, K.; Ladha, F.A.; Pettinato, A.M.; Hinson, T.J. Reading Frame Repair of TTN Truncation Variants Restores Titin Quantity and Functions. Circulation 2021, 145, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Atmanli, A.; Chai, A.C.; Cui, M.; Wang, Z.; Nishiyama, T.; Bassel-Duby, R.; Olson, E.N. Cardiac Myoediting Attenuates Cardiac Abnormalities in Human and Mouse Models of Duchenne Muscular Dystrophy. Circ. Res. 2021, 129, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, S.; Stone, O.A.; Lessard, S.; Lavertu, A.; Desjardins, J.; Beaudoin, M.; Rivas, M.; Stainier, D.Y.R.; Lettre, G. Frameshift indels introduced by genome editing can lead to in-frame exon skipping. PLoS ONE 2017, 12, e0178700. [Google Scholar] [CrossRef] [PubMed]
- Wilton-Clark, H.; Yokota, T. Antisense and Gene Therapy Options for Duchenne Muscular Dystrophy Arising from Mutations in the N-Terminal Hotspot. Genes 2022, 13, 257. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ann, F.; et al. In Vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Rivera, R.M.C.; Madhavan, S.; Pan, X.; Ann, F. In Vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Yang, Y.; Sompallae, R.; Xu, H.; Tompkins, V.S.; Holman, C.; Hose, D.; Goldschmidt, H.; Tricot, G.; Janz, S.; et al. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef]
- Fomin, A.; Gärtner, A.; Cyganek, L.; Tiburcy, M.; Tuleta, I.; Wellers, L.; Folsche, L.; Hobbach, A.J.; von Frieling-Salewsky, M.; Unger, A.; et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci. Transl. Med. 2021, 13, eabd3079. [Google Scholar] [CrossRef]
- Tiburcy, M.; Hudson, J.E.; Balfanz, P.; Schlick, S.; Meyer, T.; Liao, M.L.C.; Levent, E.; Raad, F.; Zeidler, S.; Wingender, E.; et al. Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation 2017, 135, 1832–1847. [Google Scholar] [CrossRef]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef] [Green Version]
- Schafer, S.; De Marvao, A.; Adami, E.; Fiedler, L.R.; Ng, B.; Khin, E.; Rackham, O.J.L.; Van Heesch, S.; Pua, C.J.; Kui, M.; et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat. Genet. 2017, 49, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magen, A.; Ast, G. The importance of being divisible by three in alternative splicing. Nucleic Acids Res. 2005, 33, 5574–5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewinter, M.M.; Granzier, H.L. Titin is a major human disease gene. Circulation 2013, 127, 938–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAfee, Q.; Chen, C.Y.; Yang, Y.; Caporizzo, M.A.; Morley, M.; Babu, A.; Jeong, S.; Brandimarto, J.; Bedi, K.C.; Flam, E.; et al. Truncated titin proteins in dilated cardiomyopathy. Sci. Transl. Med. 2021, 13, eabd7287. [Google Scholar] [CrossRef]
- Song, Y.; Lai, L.; Li, Z. Large-scale genomic deletions mediated by CRISPR/Cas9 system. Oncotarget 2017, 8, 5647. [Google Scholar] [CrossRef]
- Song, Y.; Yuan, L.; Wang, Y.; Chen, M.; Deng, J.; Lv, Q.; Sui, T.; Li, Z.; Lai, L. Efficient dual sgRNA-directed large gene deletion in rabbit with CRISPR/Cas9 system. Cell. Mol. Life Sci. 2016, 73, 2959–2968. [Google Scholar] [CrossRef]
- Shah, R.R.; Cholewa-Waclaw, J.; Davies, F.C.J.; Paton, K.M.; Chaligne, R.; Heard, E.; Abbott, C.M.; Bird, A.P. Efficient and versatile CRISPR engineering of human neurons in culture to model neurological disorders. Wellcome Open Res. 2016, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A. Titin: An Analysis of Genetic Variation and Cardiac Phenotype; Imperial College London: London, UK, 2014. [Google Scholar]
- Mout, R.; Ray, M.; Lee, Y.; Scaletti, F.; Rotello, V. In Vivo Delivery of CRISPR/Cas9 for Therapeutic Gene Editing: Progress and Challenges. Bioconjug. Chem. 2017, 28, 880–884. [Google Scholar] [CrossRef] [Green Version]
- Louadi, Z.; Yuan, K.; Gress, A.; Tsoy, O.; Kalinina, O.V.; Baumbach, J.; Kacprowski, T.; List, M. DIGGER: Exploring the functional role of alternative splicing in protein interactions. Nucleic Acids Res. 2021, 49, D309–D318. [Google Scholar] [CrossRef]
- Kontrogianni-Konstantopoulos, A.; Ackermann, M.A.; Bowman, A.L.; Yap, S.V.; Bloch, R.J. Muscle giants: Molecular scaffolds in sarcomerogenesis. Physiol. Rev. 2009, 89, 1217–1267. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.A.; Kontrogianni-Konstantopoulos, A. Cardiomyopathies: When the Goliaths of Heart Muscle Hurt. In Cardiomyopathies; Milei, J., Ambrosio, G., Eds.; IntechOpen: London, UK, 2013; pp. 63–79. [Google Scholar]
- Arimura, T.; Matsumoto, Y.; Okazaki, O.; Hayashi, T.; Takahashi, M.; Inagaki, N.; Hinohara, K.; Ashizawa, N.; Yano, K.; Kimura, A. Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2007, 362, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, A.; Koch, D.; Grover, S.; Rees, M.; Gautel, M. When is an obscurin variant pathogenic? The impact of Arg4344Gln and Arg4444Trp variants on protein-protein interactions and protein stability. Hum. Mol. Genet. 2021, 30, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Borisov, A.B.; Raeker, M.O.; Kontrogianni-Konstantopoulos, A.; Yang, K.; Kurnit, D.M.; Bloch, R.J.; Russell, M.W. Rapid response of cardiac obscurin gene cluster to aortic stenosis: Differential activation of Rho-GEF and MLCK and involvement in hypertrophic growth. Biochem. Biophys. Res. Commun. 2003, 310, 910–918. [Google Scholar] [CrossRef]
- Lam, L.T.; Holt, I.; Laitila, J.; Hanif, M.; Pelin, K.; Wallgren-Pettersson, C.; Sewry, C.A.; Morris, G.E. Two alternatively-spliced human nebulin isoforms with either exon 143 or exon 144 and their developmental regulation. Sci. Rep. 2018, 8, 15728. [Google Scholar] [CrossRef]
- Arimura, T.; Nakamura, T.; Hiroi, S.; Satoh, M.; Takahashi, M.; Ohbuchi, N.; Ueda, K.; Nouchi, T.; Yamaguchi, N.; Akai, J.; et al. Characterization of the human nebulette gene: A polymorphism in an actin-binding motif is associated with nonfamilial idiopathic dilated cardiomyopathy. Hum. Genet. 2000, 107, 440–451. [Google Scholar] [CrossRef]
- Purevjav, E.; Varela, J.; Morgado, M.; Kearney, D.L.; Li, H.; Taylor, M.D.; Arimura, T.; Moncman, C.L.; McKenna, W.; Murphy, R.T.; et al. Nebulette Mutations are Associated with Dilated Cardiomyopathy and Endocardial Fibroelastosis. J. Am. Coll. Cardiol. 2010 2010, 56, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Perrot, A.; Tomasov, P.; Villard, E.; Faludi, R.; Melacini, P.; Lossie, J.; Lohmann, N.; Richard, P.; De Bortoli, M.; Angelini, A.; et al. Mutations in NEBL encoding the cardiac Z-disk protein nebulette are associated with various cardiomyopathies. Arch. Med. Sci. 2016, 12, 263–278. [Google Scholar] [CrossRef]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of Human-Induced Pluripotent Stem Cells. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef]
- Kolanowski, T.J.; Antos, C.L.; Guan, K. Making human cardiomyocytes up to date: Derivation, maturation state and perspectives. Int. J. Cardiol. 2017, 241, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Zhang, Y.; Song, L.; Luo, J.; Qi, W.; Hu, J.; Lu, D.; Yang, Z.; Zhang, J. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Res. 2016, 26, 1099–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, A.K.; Molenaar, B.; Versteeg, D.; Leitoguinho, A.R.; Demkes, C.; Spanjaard, B.; De Ruiter, H.; Moqadam, F.A.; Kooijman, L.; Zentilin, L.; et al. Postnatal Cardiac Gene Editing Using CRISPR/Cas9 With AAV9-Mediated Delivery of Short Guide RNAs Results in Mosaic Gene Disruption. Circ. Res. 2017, 121, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Gori, J.L.; Hsu, P.D.; Maeder, M.L.; Shen, S.; Welstead, G.G.; Bumcrot, D. Delivery and Specificity of CRISPR/Cas9 Genome Editing Technologies for Human Gene Therapy. Hum. Gene Ther. 2015, 26, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoudian-Sani, M.R.; Farnoosh, G.; Mahdavinezhad, A.; Saidijam, M. CRISPR genome editing and its medical applications. Biotechnol. Biotechnol. Equip. 2018, 32, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In Vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef] [Green Version]
- Lai, N.C.; Gao, M.H.; Giamouridis, D.; Suarez, J.; Miyanohara, A.; Parikh, J.; Hightower, S.; Guo, T.; Dillmann, W.; Kim, Y.-C.; et al. Intravenous AAV8 Encoding Urocortin-2 Increases Function of the Failing Heart in Mice. Hum. Gene Ther. 2015, 26, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Bish, L.T.; Morine, K.; Sleeper, M.M.; Sanmiguel, J.; Wu, D.; Gao, G.; Wilson, J.M.; Sweeney, H.L. Adeno-Associated Virus (AAV) Serotype 9 Provides Global Cardiac Gene Transfer Superior to AAV1, AAV6, AAV7, and AAV8 in the Mouse and Rat. Hum. Gene Ther. 2008, 19, 1359–1368. [Google Scholar] [CrossRef]
- Chakrabarti, A.M.; Henser-Brownhill, T.; Monserrat, J.; Poetsch, A.R.; Luscombe, N.M.; Scaffidi, P. Target-specific precision of CRISPR-mediated genome editing. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Sakharkar, M.K.; Chow, V.T.K.; Kangueane, P. Distributions of exons and introns in the human genome. In Silico Biol. 2004, 4, 387–393. [Google Scholar]
- McGuigan, K.; Phillips, P.C.; Postlethwait, J.H. Evolution of sarcomeric myosin heavy chain genes: Evidence from fish. Mol. Biol. Evol. 2004, 21, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wu, F.; Mosenson, J.; Zhang, H.; He, T.C.; Wu, W.S. CRISPR/Cas9-Mediated Genome Editing Corrects Dystrophin Mutation in Skeletal Muscle Stem Cells in a Mouse Model of Muscle Dystrophy. Mol. Ther. Nucleic Acids 2017, 7, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Amoasii, L.; Mireault, A.A.; Mcanally, J.R. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [Green Version]
- Le Hir, M.; Goyenvalle, A.; Peccate, C.; Précigout, G.; Davies, K.E.; Voit, T.; Garcia, L.; Lorain, S. AAV Genome Loss From Dystrophic Mouse Muscles Therapy. Mol. Ther. 2013, 21, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Carmignac, V.; Salih, M.A.M.; Quijano-Roy, S.; Marchand, S.; Al Rayess, M.M.; Mukhtar, M.M.; Urtizberea, J.A.; Labeit, S.; Guicheney, P.; Leturcq, F.; et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann. Neurol. 2007, 61, 340–351. [Google Scholar] [CrossRef]
- Chauveau, C.; Bonnemann, C.G.; Julien, C.; Kho, A.L.; Marks, H.; Talim, B.; Maury, P.; Arne-Bes, M.C.; Uro-Coste, E.; Hu, N.; et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum. Mol. Genet. 2014, 23, 980–991. [Google Scholar] [CrossRef]
- Nugraha, B.; Buono, M.F.; von Boehmer, L.; Hoerstrup, S.P.; Emmert, M.Y. Human Cardiac Organoids for Disease Modeling. Clin. Pharmacol. Ther. 2019, 105, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.J.S.; Ang, Y.S.; Fu, J.D.; Rivas, R.N.; Mohamed, T.M.A.; Higgs, G.C.; Srivastava, D.; Pruitt, B.L. Contractility of Single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc. Natl. Acad. Sci. USA 2015, 112, 12705–12710. [Google Scholar] [CrossRef] [Green Version]
- Blazeski, A.; Lowenthal, J.; Wang, Y.; Teuben, R.; Zhu, R.; Gerecht, S.; Tomaselli, G.; Tung, L. Engineered Heart Slice Model of Arrhythmogenic Cardiomyopathy Using Plakophilin-2 Mutant Myocytes. Tissue Eng. Part A 2019, 25, 725–735. [Google Scholar] [CrossRef]
- Savarese, M.; Sarparanta, J.; Vihola, A.; Udd, B.; Hackman, P. Increasing Role of Titin Mutations in Neuromuscular Disorders. J. Neuromuscul. Dis. 2016, 3, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]

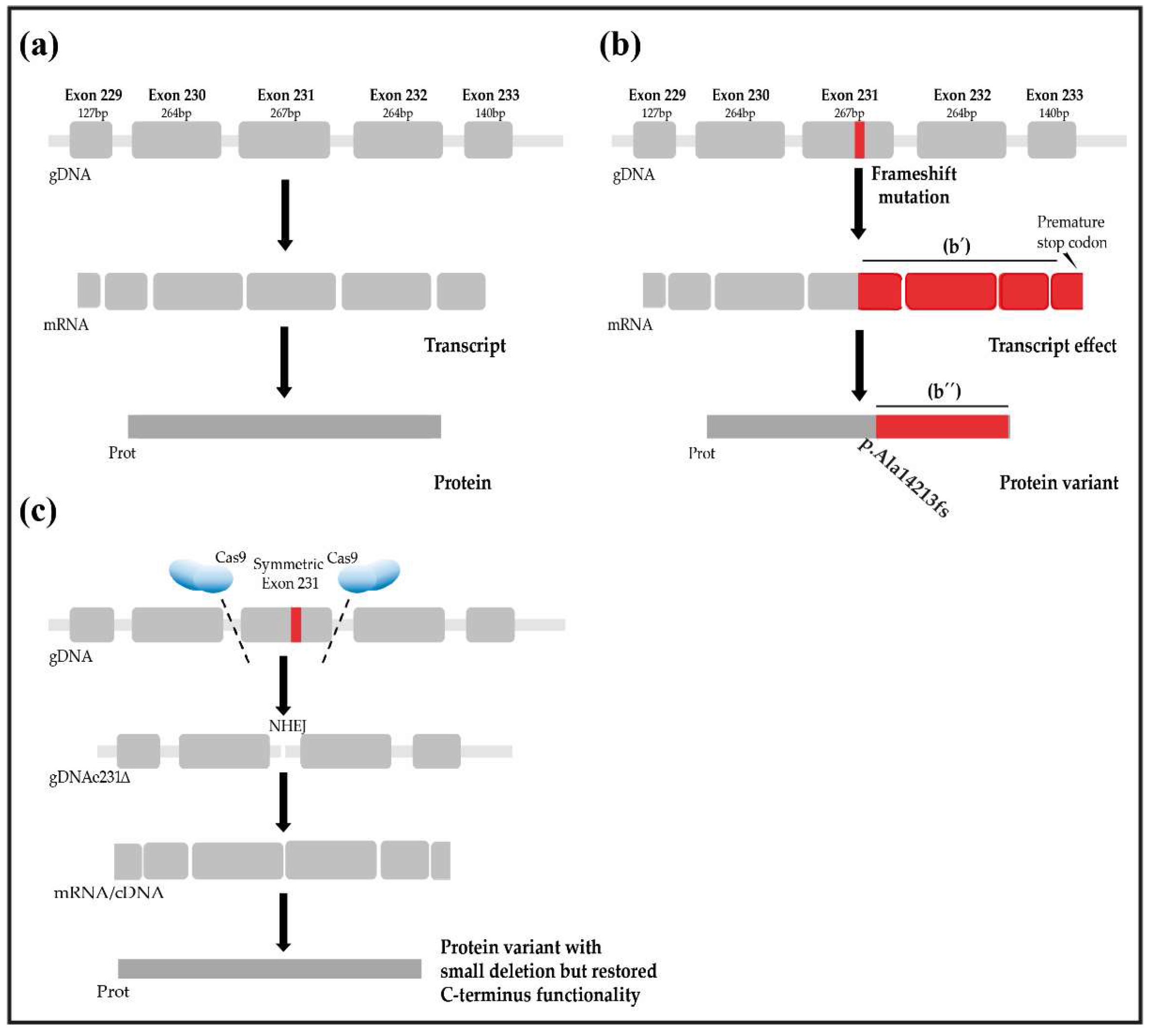
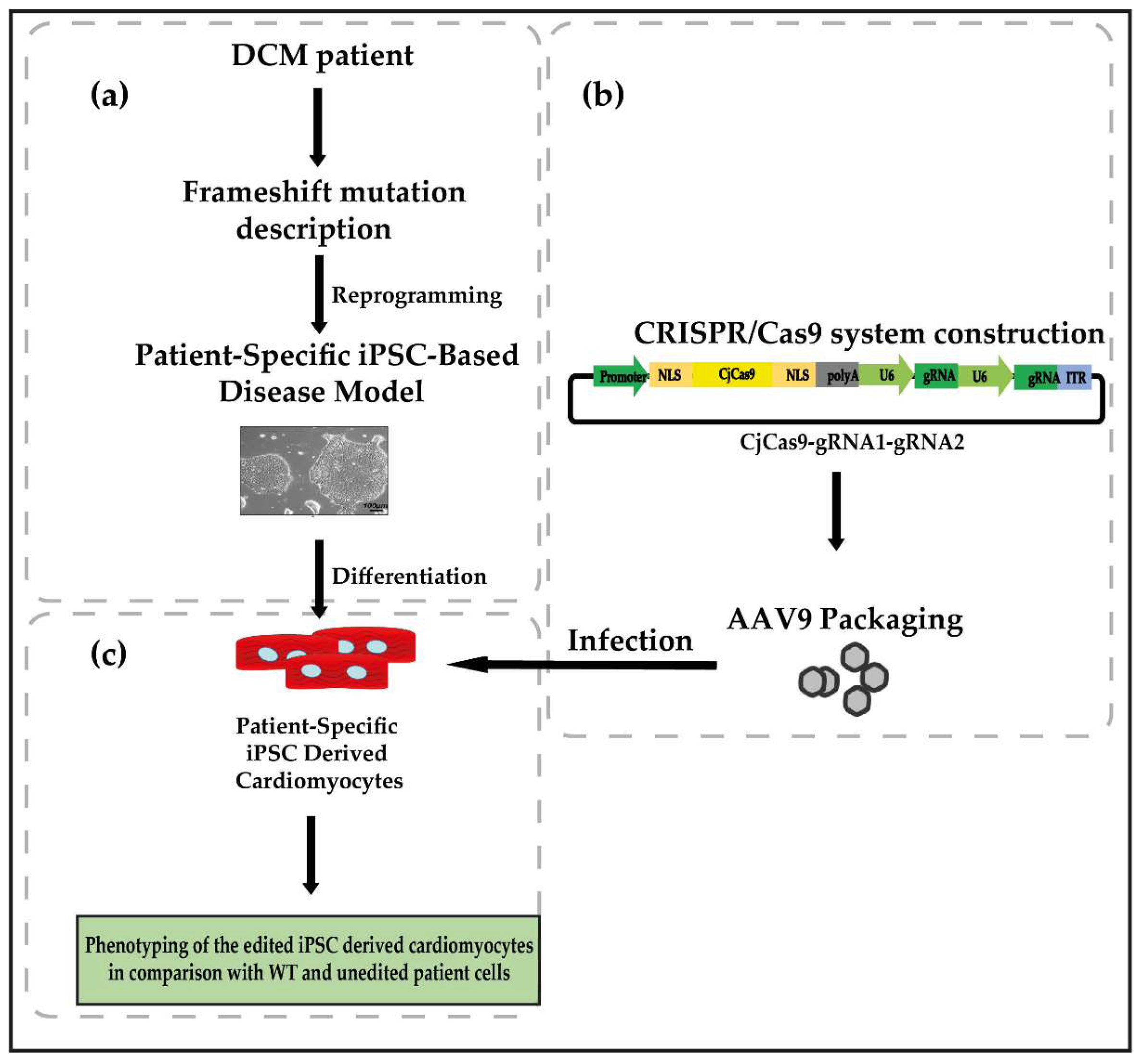
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Polo, I.; Behr, R. Exploring the Potential of Symmetric Exon Deletion to Treat Non-Ischemic Dilated Cardiomyopathy by Removing Frameshift Mutations in TTN. Genes 2022, 13, 1093. https://doi.org/10.3390/genes13061093
Rodriguez-Polo I, Behr R. Exploring the Potential of Symmetric Exon Deletion to Treat Non-Ischemic Dilated Cardiomyopathy by Removing Frameshift Mutations in TTN. Genes. 2022; 13(6):1093. https://doi.org/10.3390/genes13061093
Chicago/Turabian StyleRodriguez-Polo, Ignacio, and Rüdiger Behr. 2022. "Exploring the Potential of Symmetric Exon Deletion to Treat Non-Ischemic Dilated Cardiomyopathy by Removing Frameshift Mutations in TTN" Genes 13, no. 6: 1093. https://doi.org/10.3390/genes13061093
APA StyleRodriguez-Polo, I., & Behr, R. (2022). Exploring the Potential of Symmetric Exon Deletion to Treat Non-Ischemic Dilated Cardiomyopathy by Removing Frameshift Mutations in TTN. Genes, 13(6), 1093. https://doi.org/10.3390/genes13061093