
The Impact of SKP2 Gene Expression in Chronic Myeloid Leukemia

 , , ,
, , ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Peripheral Blood Collection
2.2. Molecular Analysis of BCR–ABL1 Transcript
2.3. Mononuclear Cell Isolation, RNA Extraction, and cDNA Synthesis for SKP2 Gene Expression Analysis
2.4. Real-Time Reverse Transcriptase–Polymerase Chain Reaction (RT-PCR)
2.5. SKP2 Protein Level Assay
2.6. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
6. Limitations of the Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heisterkamp, N.; Stam, K.; Groffen, J.; de Klein, A.; Grosveld, G. Structural organization of the bcr gene and its role in the Ph’ translocation. Nature 1985, 315, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Daley, G.Q.; Van Etten, R.A.; Baltimore, D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990, 247, 824–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehlmann, R.; Hochhaus, A.; Baccarani, M.; European, L. Chronic myeloid leukaemia. Lancet 2007, 370, 342–350. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Antolini, L.; Mahon, F.X.; Guilhot, F.; Deininger, M.; Fava, C.; Nagler, A.; Maria Della Casa, C.; Morra, E.; Abruzzese, E.; et al. Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J. Natl. Cancer Inst. 2011, 103, 553–561. [Google Scholar] [CrossRef]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic myeloid leukemia: Mechanisms of blastic transformation. J. Clin. Investig. 2010, 120, 2254–2264. [Google Scholar] [CrossRef] [Green Version]
- Gambacorti-Passerini, C.B.; Gunby, R.H.; Piazza, R.; Galietta, A.; Rostagno, R.; Scapozza, L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 2003, 4, 75–85. [Google Scholar] [CrossRef]
- Mughal, T.I.; Radich, J.P.; Deininger, M.W.; Apperley, J.F.; Hughes, T.P.; Harrison, C.J.; Gambacorti-Passerini, C.; Saglio, G.; Cortes, J.; Daley, G.Q. Chronic myeloid leukemia: Reminiscences and dreams. Haematologica 2016, 101, 541–558. [Google Scholar] [CrossRef]
- O’Hare, T.; Eide, C.A.; Deininger, M.W. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249. [Google Scholar] [CrossRef]
- Mathisen, M.S.; Kantarjian, H.M.; Cortes, J.; Jabbour, E. Mutant BCR-ABL clones in chronic myeloid leukemia. Haematologica 2011, 96, 347–349. [Google Scholar] [CrossRef] [Green Version]
- Hochhaus, A.; Kreil, S.; Corbin, A.S.; La Rosée, P.; Müller, M.C.; Lahaye, T.; Hanfstein, B.; Schoch, C.; Cross, N.C.P.; Berger, U.; et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002, 16, 2190–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahon, F.X.; Belloc, F.; Lagarde, V.; Chollet, C.; Moreau-Gaudry, F.; Reiffers, J.; Goldman, J.M.; Melo, J.V. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 2003, 101, 2368–2373. [Google Scholar] [CrossRef]
- Rosti, G.; Castagnetti, F.; Gugliotta, G.; Baccarani, M. Tyrosine kinase inhibitors in chronic myeloid leukaemia: Which, when, for whom? Nat. Rev. Clin. Oncol. 2017, 14, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A phase 2 trial of ponatinib in Philadelphia chromosomepositive leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pophali, P.A.; Patnaik, M.M. The role of new tyrosine kinase inhibitors in chronic myeloid leukemia. Cancer, J. 2016, 22, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Pearson, K.; Ferguson, J.E.; Clark, R.E. The early molecular response to imatinib predicts cytogenetic and clinical outcome in chronic myeloid leukaemia. Br. J. Haematol. 2003, 120, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Marin, D.; Ibrahim, A.R.; Lucas, C.; Gerrard, G.; Wang, L.; Szydlo, R.M.; Clark, R.E.; Apperley, J.F.; Milojkovic, D.; Bua, M.; et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J. Clin. Oncol. 2012, 30, 232–238. [Google Scholar] [CrossRef]
- Hanfstein, B.; Müller, M.C.; Hehlmann, R.; Erben, P.; Lauseker, M.; Fabarius, A.; Schnittger, S.; Haferlach, C.; Göhring, G.; Proetel, U.; et al. Early molecular and cytogenetic response is predictive for long-term progression-free and overall survival in chronic myeloid leukemia (CML). Leukemia 2012, 26, 2096–2102. [Google Scholar] [CrossRef] [Green Version]
- Branford, S.; Kim, D.-W.; Soverini, S.; Haque, A.; Shou, Y.; Woodman, R.C.; Kantarjian, H.M.; Martinelli, G.; Radich, J.P.; Saglio, G.; et al. Initial molecular response at 3 months may predict both response and event-free survival at 24 months in imatinib-resistant or -intolerant patients with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase treated with nilotinib. J. Clin. Oncol. 2012, 30, 4323–4329. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Kantarjian, H.M.; Saglio, G.; Steegmann, J.L.; Shah, N.P.; Boqué, C.; Chuah, C.; Pavlovsky, C.; Mayer, J.; Cortes, J.; et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood 2014, 123, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Hanfstein, B.; for the SAKK and the German CML Study Group; Shlyakhto, V.; Lauseker, M.; Hehlmann, R.; Saussele, S.; Dietz, C.; Erben, P.; Fabarius, A.; Proetel, U.; et al. Velocity of early BCR-ABL transcript elimination as an optimized predictor of outcome in chronic myeloid leukemia (CML) patients in chronic phase on treatment with imatinib. Leukemia 2014, 28, 1988–1992. [Google Scholar] [CrossRef] [PubMed]
- Branford, S.; Yeung, D.T.; Parker, W.T.; Roberts, N.; Purins, L.; Braley, J.A.; Altamura, H.K.; Yeoman, A.L.; Georgievski, J.; Jamison, B.A.; et al. Prognosis for patients with CML and 10% BCR-ABL1 after 3 months of imatinib depends on the rate of BCR-ABL1 decline. Blood 2014, 124, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Suresh, B.; Lee, J.; Kim, H.; Ramakrishna, S. Regulation of pluripotency and differentiation by deubiquitinating enzymes. Cell Death Differ. 2016, 23, 1257–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfoh, R.; Lacdao, I.K.; Saridakis, V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr.-Relat. Cancer 2015, 22, T35–T54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating enzymes in cellular signaling and disease regulation. IUBMB Life 2015, 67, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Yang, L.; Wang, G.; Ding, G.; Peng, B.; Wen, Y.; Wang, Z. Inhibition of Skp2 sensitizes lung cancer cells to paclitaxel. Onco Targets Ther. 2017, 10, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.S.; Adamson, A.; Lin, Z.; Perez-Ordonez, B.; Jordan, R.C.K.; Tripp, S.; Perkins, S.L.; Elenitoba-Johnson, K.S.J. Expression of Skp2, a p27 (Kip1) ubiquitin ligase, in malignant lymphoma: Correlation with p27 (Kip1) and proliferation index. Blood 2002, 100, 2950–2956. [Google Scholar] [CrossRef]
- Wang, G.; Chan, C.-H.; Gao, Y.; Lin, H.K. Novel roles of Skp2 E3 ligase in cellular senescence, cancer progression, and metastasis. Chin. J. Cancer 2012, 31, 169–177. [Google Scholar] [CrossRef]
- Zhang, W.; Cao, L.; Sun, Z.; Xu, J.; Tang, L.; Chen, W.; Luo, J.; Yang, F.; Wang, Y.; Guan, X. Skp2 is over-expressed in breast cancer and promotes breast cancer cell proliferation. Cell Cycle 2016, 15, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Bu, W.; Luo, T. miR-1297 Promotes Cell Proliferation of Non-Small Cell Lung Cancer Cells: Involving in PTEN/Akt/Skp2 Signaling Pathway. DNA Cell Biol. 2017, 36, 976–982. [Google Scholar] [CrossRef]
- Su, J.; Zhou, X.; Wang, L.; Yin, X.; Wang, Z. Curcumin inhibits cell growth and invasion and induces apoptosis through down-regulation of Skp2 in pancreatic cancer cells. Am. J. Cancer Res. 2016, 6, 1949–1962. [Google Scholar] [PubMed]
- Wen, Y.; Yang, K.; Wang, K. Inhibiting the role of Skp2 suppresses cell proliferation and tumorigenesis of human gastric cancer cells via the upregulation of p27kip1. Mol. Med. Rep. 2016, 14, 3917–3924. [Google Scholar] [CrossRef] [Green Version]
- Arbini, A.A.; Greco, M.; Yao, J.L.; Bourne, P.; Marra, E.; Hsieh, J.T.; di Sant’agnese, P.A.; Moro, L. Skp2 overexpression is associated with loss of BRCA2 protein in human prostate cancer. Am. J. Pathol. 2011, 178, 2367–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woenckhaus, C.; Maile, S.; Uffmann, S.; Bansemir, M.; Dittberner, T.; Poetsch, M.; Giebel, J. Expression of Skp2 and p27KIP1 in naevi and malignant melanoma of the skin and its relation to clinical outcome. Histol. Histopathol. 2005, 20, 501–508. [Google Scholar] [PubMed]
- Yang, Y.; Yan, W.; Liu, Z.; Wei, M. Skp2 inhibitor SKPin C1 decreased viability and proliferation of multiple myeloma cells and induced apoptosis. Braz. J. Med. Biol. Res. 2019, 52, e8412. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Yang, Y.; Yang, W. Inhibition of SKP2 Activity Impaired ATM-Mediated DNA Repair and Enhanced Sensitivity of Cisplatin-Resistant Mantle Cell Lymphoma Cells. Cancer Biother. Radiopharm. 2019, 34, 451–458. [Google Scholar] [CrossRef]
- Chen, X.; Huang, Z.; Wu, W.; Xia, R. Inhibition of Skp2 Sensitizes Chronic Myeloid Leukemia Cells to Imatinib. Cancer Manag. Res. 2020, 12, 4777–4787. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, N.; Xia, X.; Guo, Z.; Li, Y.; Jiang, L.; Zhou, R.; Tang, D.; Huang, H.; Liu, J. USP10 modulates the SKP2/Bcr-Abl axis via stabilizing SKP2 in chronic myeloid leukemia. Cell Discov. 2019, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef]
- Sokal, J.E.; Cox, E.B.; Baccarani, M.; Tura, S.; Gomez, G.A.; Robertson, J.E.; Tso, C.Y.; Braun, T.J.; Clarkson, B.D.; Cervantes, F.; et al. Prognostic discrimination in “good risk” chronic granulocytic leukemia. Blood 1984, 63, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Castagnetti, F.; Gugliotta, G.; Breccia, M.; Stagno, F.; Iurlo, A.; Albano, F.; Abruzzese, E.; Martino, B.; Levato, L.; Intermesoli, T.; et al. Long-term outcome of chronic myeloid leukemia patients treated frontline with imatinib. Leukemia 2015, 29, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.Z.; Liu, Y.R.; Zhu, H.H.; Li, J.L.; Ruan, G.R.; Zhang, Y.; Jiang, Q.; Jiang, H.; Li, L.D.; Chang, Y.; et al. Different kinetic patterns of BCR-ABL transcript levels in imatinib-treated chronic myeloid leukemia patients after achieving complete cytogenetic response. Int. J. Lab. Hematol. 2008, 30, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C. Standardisation of molecular monitoring for chronic myeloid leukaemia. Best Pract. Res. Clin. Haematol. 2009, 22, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Branford, S.; Cross, N.; Hochhaus, A.; Radich, J.; Saglio, G.; Kaeda, J.; Goldman, J.; Hughes, T. Rationale for the recommendations for harmonizing current methodology for detecting BCR-ABL transcripts in patients with chronic myeloid leukaemia. Leukemia 2006, 20, 1925–1930. [Google Scholar] [CrossRef]
- Branford, S.; Fletcher, L.; Cross, N.; Müller, M.C.; Hochhaus, A.; Kim, D.-W.; Radich, J.P.; Saglio, G.; Pane, F.; Kamel-Reid, S.; et al. Desirable performance characteristics for BCR-ABL measurement on an international reporting scale to allow consistent interpretation of individual patient response and comparison of response rates between clinical trials. Blood 2008, 112, 3330–3338. [Google Scholar] [CrossRef]
- White, H.E.; Matejtschuk, P.; Rigsby, P.; Gabert, J.; Lin, F.; Wang, Y.L.; Branford, S.; Müller, M.C.; Beaufils, N.; Beillard, E.; et al. Establishment of the first World Health Organization International Genetic Reference Panel for quantitation of BCR-ABL mRNA. Blood 2010, 116, e111–e117. [Google Scholar] [CrossRef] [Green Version]
- White, H.E.; Hedges, J.; Bendit, I.; Branford, S.; Colomer, D.; Hochhaus, A.; Hughes, T.; Kamel-Reid, S.; Kim, D.-W.; Modur, V.; et al. Establishment and validation of analytical reference panels for the standardization of quantitative BCR-ABL1 measurements on the international scale. Clin. Chem. 2013, 59, 938–948. [Google Scholar] [CrossRef]
- Nowell, P.C.; Hungerford, D.A. A minute chromosome in human chronic granulocytic leukemia. J. Natl. Cancer Inst. 1960, 25, 85. [Google Scholar]
- Caspersson, T.; Gahrton, G.; Lindsten, J.; Zech, L. Identification of the Philadelphia chromosome as a number 22 by quinacrine mustard fluorescence analysis. Exp. Cell Res. 1970, 63, 238. [Google Scholar] [CrossRef]
- Bartram, C.R.; de Klein, A.; Hagemeijer, A.; van Agthoven, T.; van Kessel, A.G.; Bootsma, D.; Grosveld, G.; Ferguson-Smith, M.A.; Davies, T.; Stone, M.; et al. Translocation of c-abl oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukemia. Nature 1983, 306, 277. [Google Scholar] [CrossRef]
- Jabbour, E.; Branford, S.; Saglio, G.; Jones, D.; Cortes, J.E.; Kantarjian, H.M. Practical advice for determining the role of BCR-ABL mutations in guiding tyrosine kinase inhibitor therapy in patients with chronic myeloid leukemia. Cancer 2011, 117, 1800–1811. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Xu, N.; Zhou, X.; Huang, J.; Wan-Er, W.; Chen, C.; Liang, L.; Liu, Q.; Xiaoli, L. PKM2 mediates chronic myeloid leukemia Imatinib resistance by regulating glycolysis energy metabolism. Blood 2018, 132, 1724. [Google Scholar] [CrossRef]
- Yang, Q.; Huang, J.; Wu, Q.; Cai, Y.; Zhu, L.; Lu, X.; Chen, S.; Chen, C.; Wang, Z. Acquisition of epithelial-mesenchymal transition is associated with Skp2 expression in paclitaxel-resistant breast cancer cells. Br. J. Cancer 2014, 110, 1958–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Q.; Jiang, Z.; Li, Y.; Wu, H.; Yu, J.; Jiang, M. FAM60A promotes cisplatin resistance in lung cancer cells by activating SKP2 expression. Anticancer. Drugs 2020, 31, 776–784. [Google Scholar]
- Bhatt, S.; Stender, J.D.; Joshi, S.; Wu, G.; Katzenellenbogen, B.S. OCT-4: A novel estrogen receptor--α collaborator that promotes tamoxifen resistance in breast cancer cells. Oncogene 2016, 35, 5722–5734. [Google Scholar] [CrossRef]
- Ding, L.; Wang, C.; Cui, Y.; Han, X.; Zhou, Y.; Bai, J.; Li, R. S-phase kinase-associated protein 2 is involved in epithelialmesenchymal transition in methotrexate-resistant osteosarcoma cells. Int. J. Oncol. 2018, 52, 1841–1852. [Google Scholar]
- Donato, N.J.; Wu, J.Y.; Stapley, J.; Lin, H.; Arlinghaus, R.; Aggarwal, B.B.; Shishodin, S.; Albitar, M.; Hayes, K.; Kantarjian, H.; et al. Imatinib mesylate resistance through BCR-ABL independence in chronic myelogenous leukemia. Cancer Res. 2004, 64, 672–677. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Shan, Y.; Bai, R.; Xue, L.; Eide, C.A.; Ou, J.; Zhu, L.J.; Hutchinson, L.; Cerny, J.; Khoury, H.J.; et al. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci. Transl. Med. 2014, 6, 252ra121. [Google Scholar] [CrossRef] [Green Version]
- Roychowdhury, S.; Talpaz, M. Managing resistance in chronic myeloid leukemia. Blood Rev. 2011, 25, 279–290. [Google Scholar] [CrossRef]
- Liu, Y.; Perdreau, S.A.; Chatterjee, P.; Wang, L.; Kuan, S.F.; Duensing, A. Imatinib mesylate induces quiescence in gastrointestinal stromal tumor cells through the CDH1-SKP2-p27Kip1 signaling axis. Cancer Res. 2008, 68, 9015–9023. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yi, J.; Yoon, D.; Ryoo, Z.; Lee, I.; Kim, M. Imatinib and GNF-5 Exhibit an Inhibitory Effect on Growth of Hepatocellar Carcinoma Cells by Downregulating S-phase Kinase-associated Protein 2. J. Cancer Prev. 2020, 25, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Colarossi, S.; Gnani, A.; Rosti, G.; Castagnetti, F.; Poerio, A.; Iacobucci, I.; Amabile, M.; Abruzzese, E.; Orlandi, E.; et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: By the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin. Cancer Res. 2006, 12, 7374–7379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yang, H.; Xu, X.; Yi, S.; Meng, L. Mutations in the BCR-ABL1 kinase domain in patients with chronic myeloid leukaemia treated with TKIs or at diagnosis. Oncol. Lett. 2020, 20, 1071–1076. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CML (n = 100) | Control (n = 100) | Test of Sig. | p | |
|---|---|---|---|---|
| Age (years) | ||||
| Mean ± SD. | 54.4 ± 7.7 | 52.9 ± 7.9 | T = 1.396 | 0.164 |
| Median (Min.–Max.) | 58 (39–63) | 55.5 (39–64) | ||
| Gender | ||||
| Male | 73 (73.0%) | 70 (70.0%) | χ2 = 0.221 | 0.638 |
| Female | 27 (27.0%) | 30 (30.0%) | ||
| BCR–ABL1 IS% | ||||
| Mean ± SD. | 69.4 ± 18.2 | NA | ||
| Median (Min.–Max.) | 69.5 (33–108) | NA | NA | NA |
| SOKAL | ||||
| Low | 32 (32%) | NA | NA | NA |
| Intermediate | 41 (41%) | NA | ||
| High | 27 (27%) | NA | ||
| Relative expression of SKP2 | ||||
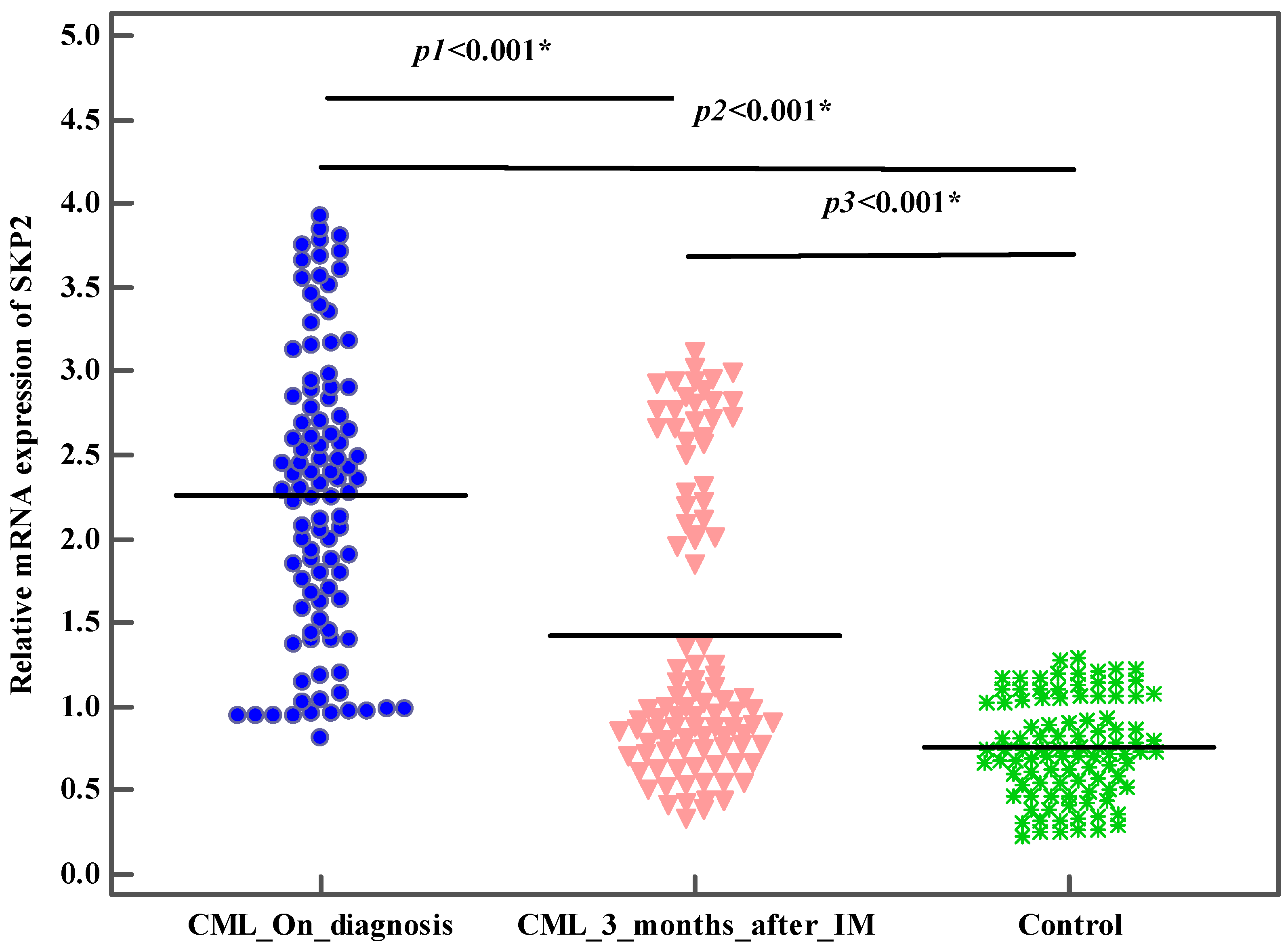
| Mean ± SD. | 2.3 ± 0.9 | 0.8 ± 0.3 | ||
| Median (Min.–Max.) | 2.3 (0.8–3.9) | 0.7 (0.2–1.3) | U = 416.50 * | <0.001 * |
| Protein levels of SKP2 (ELISA) (ng/mL) | ||||
| Mean ± SD. | 4.4 ± 1.5 | 1.2 ± 0.6 | U = 193.0 * | <0.001 * |
| Median (Min.–Max.) | 4.4 (1.2–7.6) | 1.2 (0.2–2.3) | ||
| BCR–ABL1 Mutations | ||||
| NA | 66 (66%) | NA | ||
| Yes | 12 (12%) | NA | ||
| No | 22 (22%) | NA |
| On Diagnosis | 3 Months after IM | Z | p | |
|---|---|---|---|---|
| BCR–ABL1 IS% | ||||
| Mean ± SD. | 69.4 ± 18.2 | 19.1 ± 24.7 | 8.607 * | <0.001 * |
| Median (Min.–Max.) | 69.5 (33–108) | 7 (1–90) | ||
| Relative expression of SKP2 | ||||
| Mean ± SD. | 2.3 ± 0.9 | 1.4 ± 0.9 | 8.110 * | <0.001 * |
| Median (Min.–Max.) | 2.3 (0.8–3.9) | 1 (0.3–3.1) | ||
| Protein levels of SKP2 (ELISA) (ng/mL) | ||||
| Mean ± SD. | 4.4 ± 1.5 | 2.5 ± 1.6 | 8.555 * | <0.001 * |
| Median (Min.–Max.) | 4.4 (1.2–7.6) | 2 (0.3–6.8) |
| N | SKP2 On Diagnosis | Test of Sig. | p | ||
|---|---|---|---|---|---|
| Mean ± SD | Median (Min.–Max.) | ||||
| Hematologic Response | |||||
| Yes | 96 | 2.2 ± 0.8 | 2.3 (0.8–3.9) | U = 18.0 * | <0.001 * |
| No | 4 | 3.7 ± 0.1 | 3.7 (3.6–3.8) | ||
| Cytogenetic Response | |||||
| Yes | 66 | 1.9 ± 0.7 | 1.9 (0.8–3.9) | U = 416.0 * | <0.001 * |
| No | 34 | 2.9 ± 0.7 | 3 (1.2–3.8) | ||
| Molecular Response | |||||
| BCR–ABL1 IS% < 10 | 71 | 2.0 ± 0.7 | 1.9 (0.8–3.9) | U = 331.0 * | <0.001 * |
| BCR–ABL1 IS% ≥ 10 | 29 | 3.0 ± 0.7 | 3.2 (1.2–3.8) | ||
| Treatment Response | |||||
| Optimal | 66 | 1.9 ± 0.7 | 1.9 (0.8–3.9) | U = 416.0 * | <0.001 * |
| Warning/Failure | 34 | 2.9 ± 0.7 | 3 (1.2–3.8) | ||
| BCR–ABL1 Mutations | |||||
| Yes | 12 | 2.5 ± 0.7 | 2.3 (1.2–3.7) | U = 61.0 * | 0.010 * |
| No | 22 | 3.1 ± 0.7 | 3.2 (1.2–3.8) | ||
| Crude Odds Ratio | Adjust Odds Ratio # | |||
|---|---|---|---|---|
| p | OR (95% CI) | p | OR (95% CI) | |
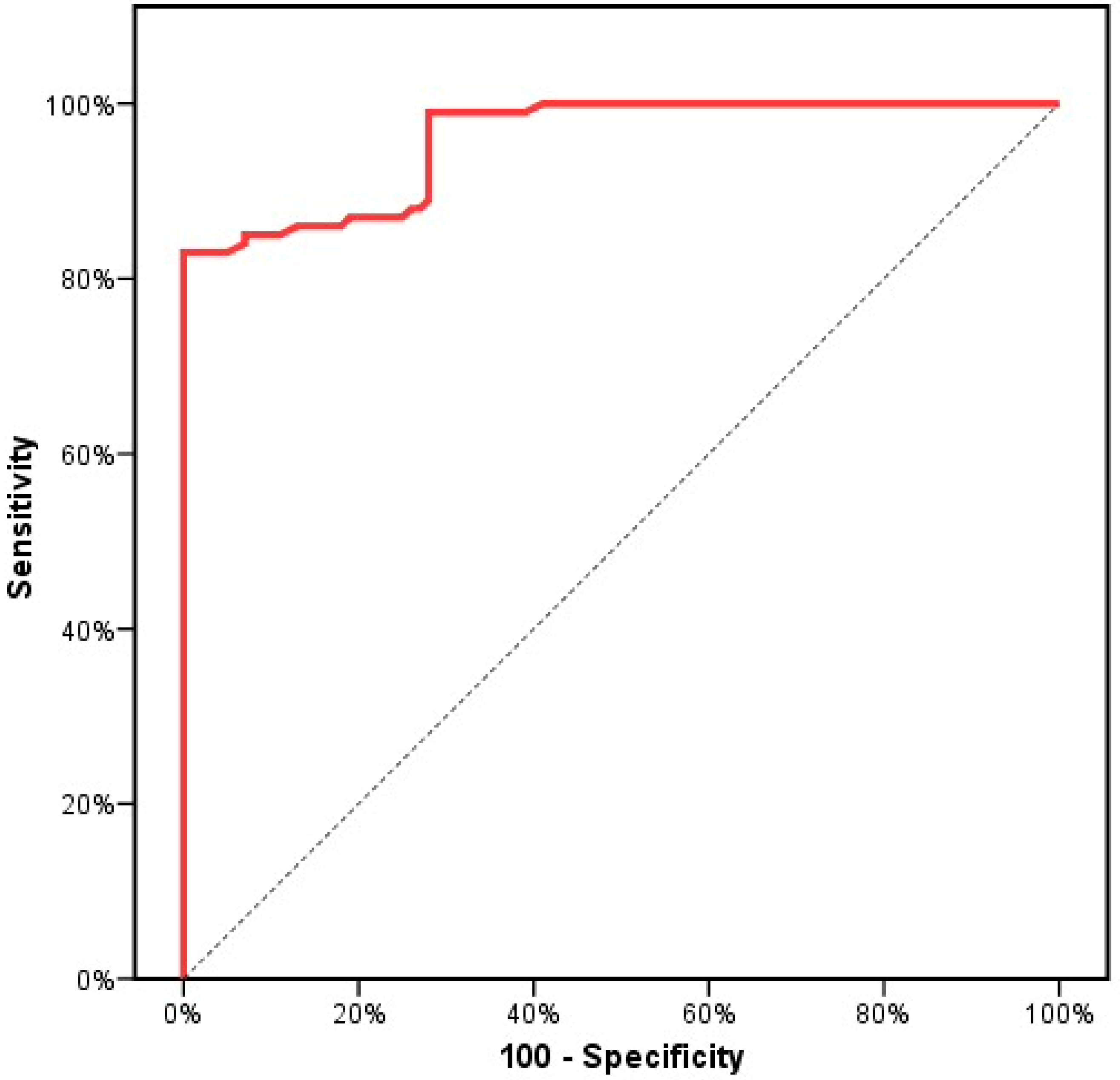
| SKP2on Diagnosis | <0.001 * | 5.259 (2.568–10.772) | <0.001 * | 5.234(2.555–10.725) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hodeib, H.; Abd EL Hai, D.; Tawfik, M.A.; Allam, A.A.; Selim, A.F.; Sarhan, M.E.; Selim, A.; Sabry, N.M.; Mansour, W.; Youssef, A. The Impact of SKP2 Gene Expression in Chronic Myeloid Leukemia. Genes 2022, 13, 948. https://doi.org/10.3390/genes13060948
Hodeib H, Abd EL Hai D, Tawfik MA, Allam AA, Selim AF, Sarhan ME, Selim A, Sabry NM, Mansour W, Youssef A. The Impact of SKP2 Gene Expression in Chronic Myeloid Leukemia. Genes. 2022; 13(6):948. https://doi.org/10.3390/genes13060948
Chicago/Turabian StyleHodeib, Hossam, Dina Abd EL Hai, Mohamed A. Tawfik, Alzahraa A. Allam, Ahmed F. Selim, Mohamed E. Sarhan, Amal Selim, Nesreen M. Sabry, Wael Mansour, and Amira Youssef. 2022. "The Impact of SKP2 Gene Expression in Chronic Myeloid Leukemia" Genes 13, no. 6: 948. https://doi.org/10.3390/genes13060948