
Genetic Alterations, DNA Methylation, Alloantibodies and Phenotypic Heterogeneity in Type III von Willebrand Disease

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Operational Definitions
2.2. Setting and Study Design
2.3. Sample Collection and Storage
2.4. Patients
2.5. Genetic Analysis
2.6. Mutational Data Analysis
2.7. Sanger Sequencing
2.8. In Silico Structural Analysis
2.9. DNA Methylation Analysis
2.10. Statistical Analysis
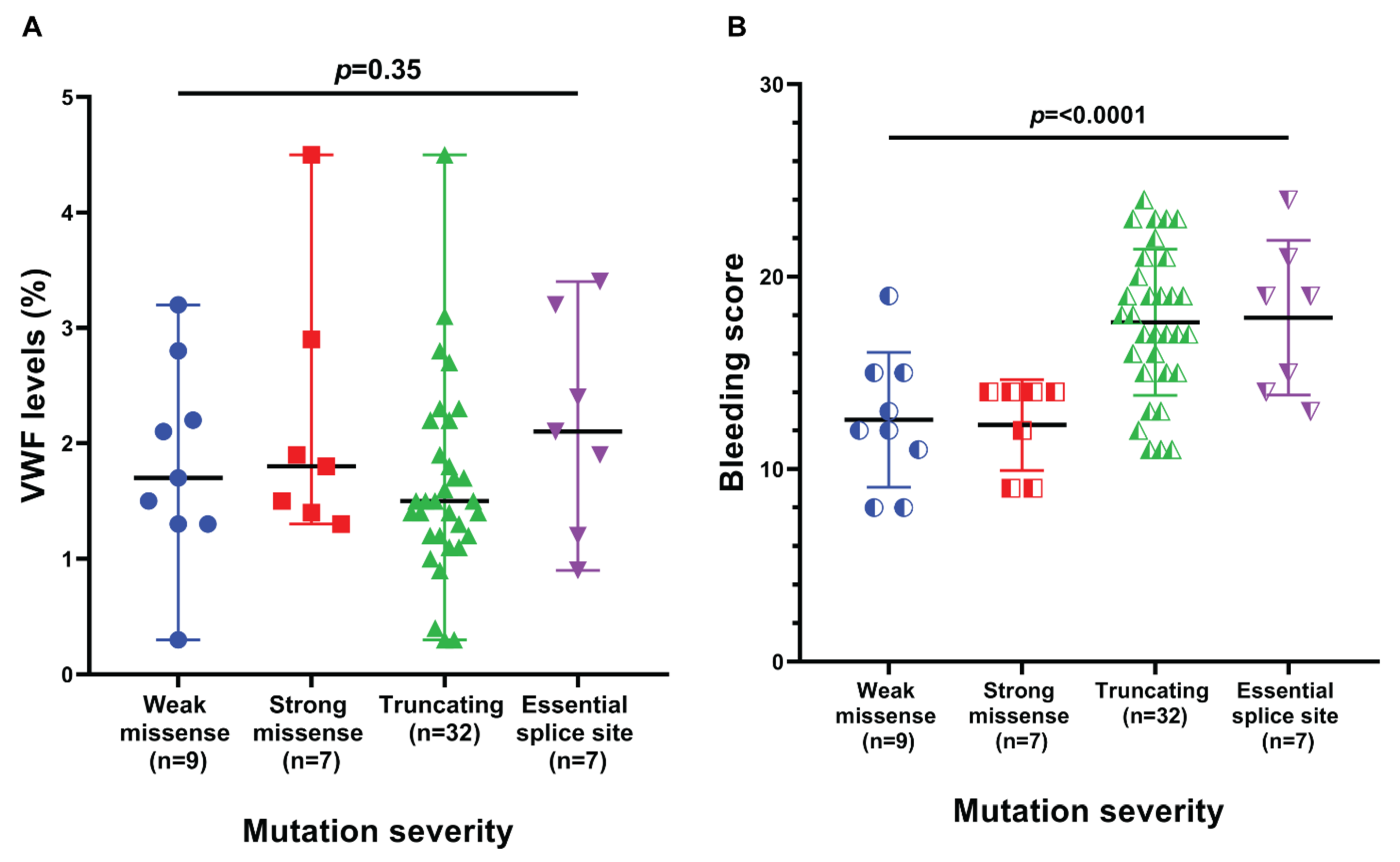
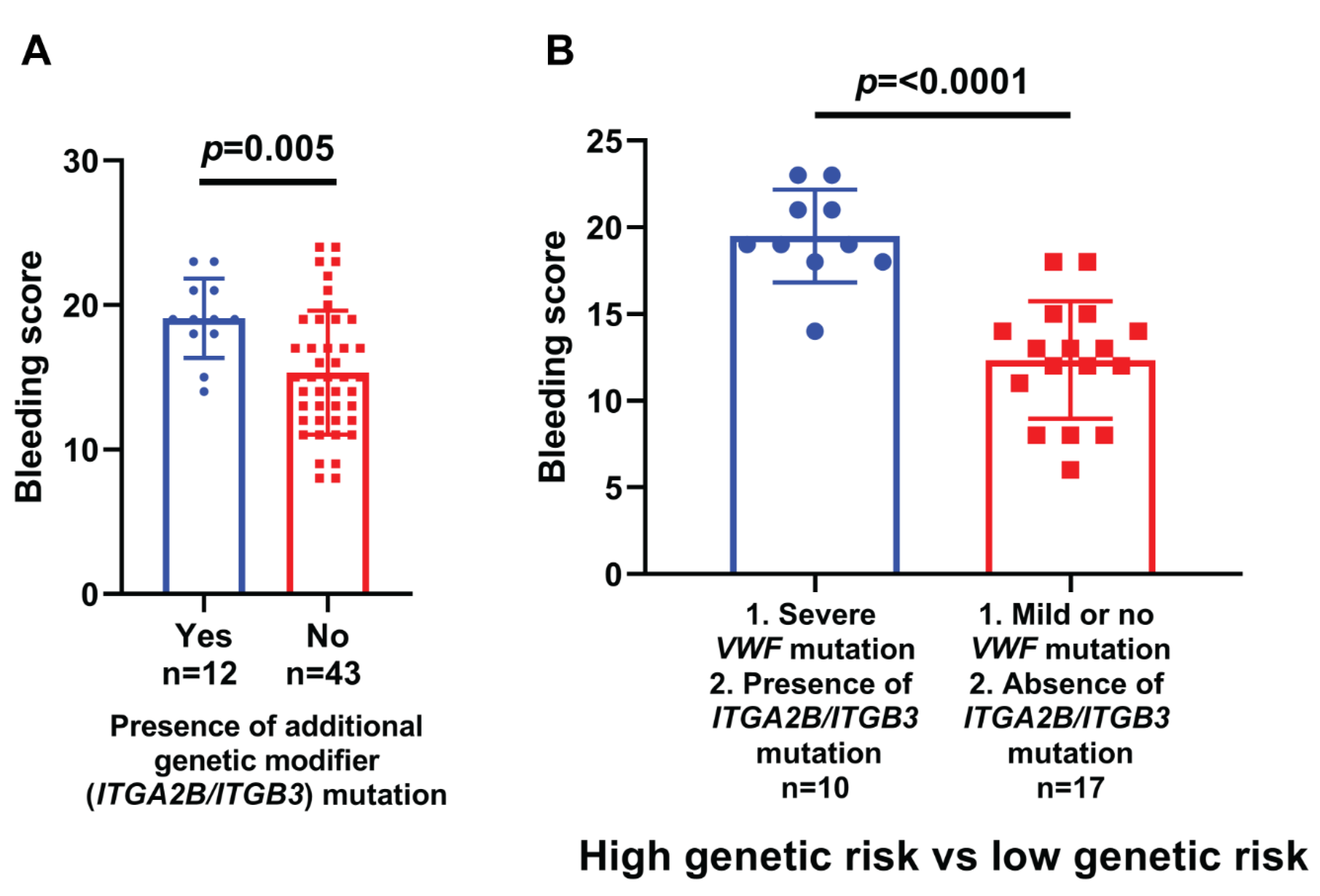
3. Results
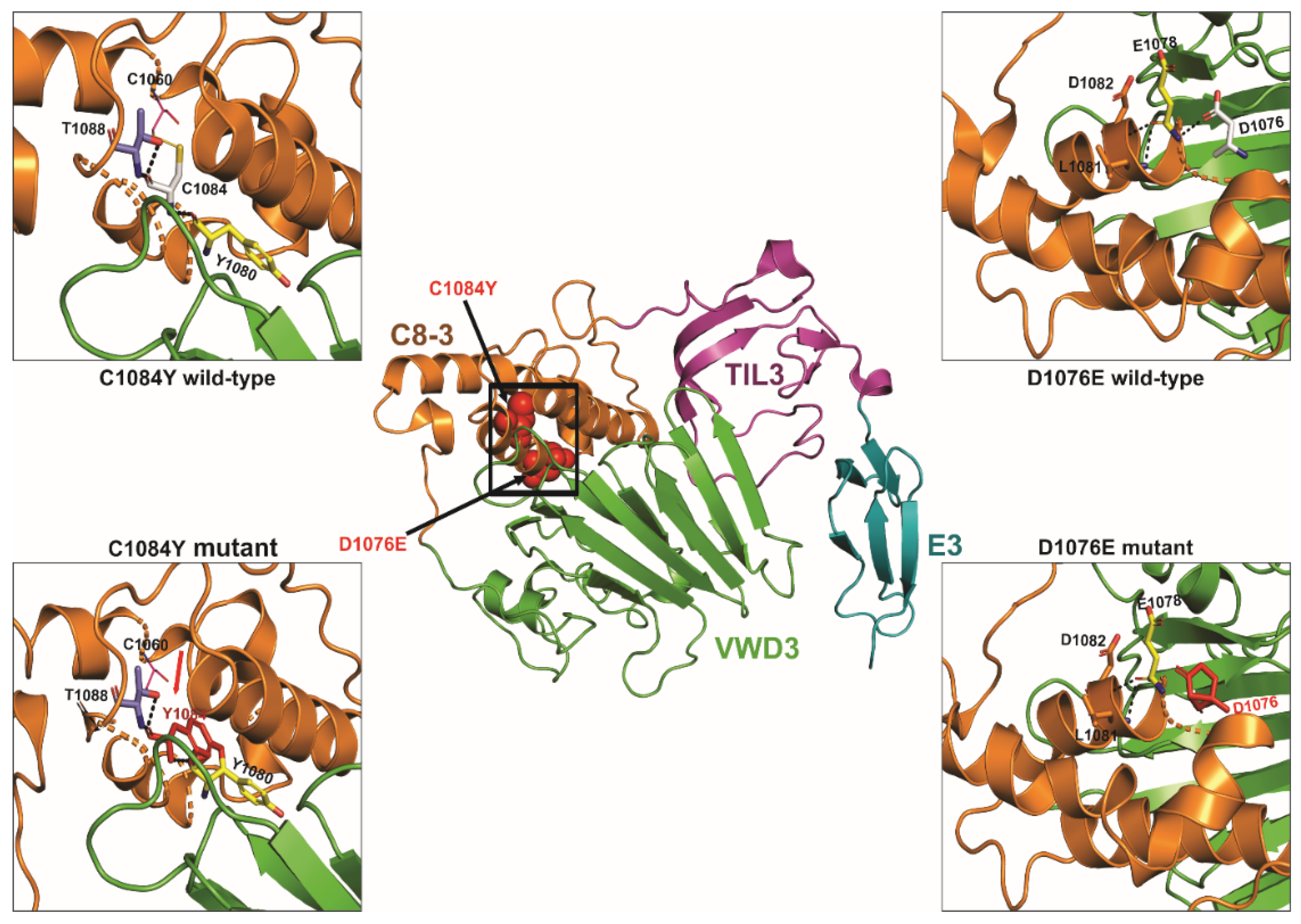
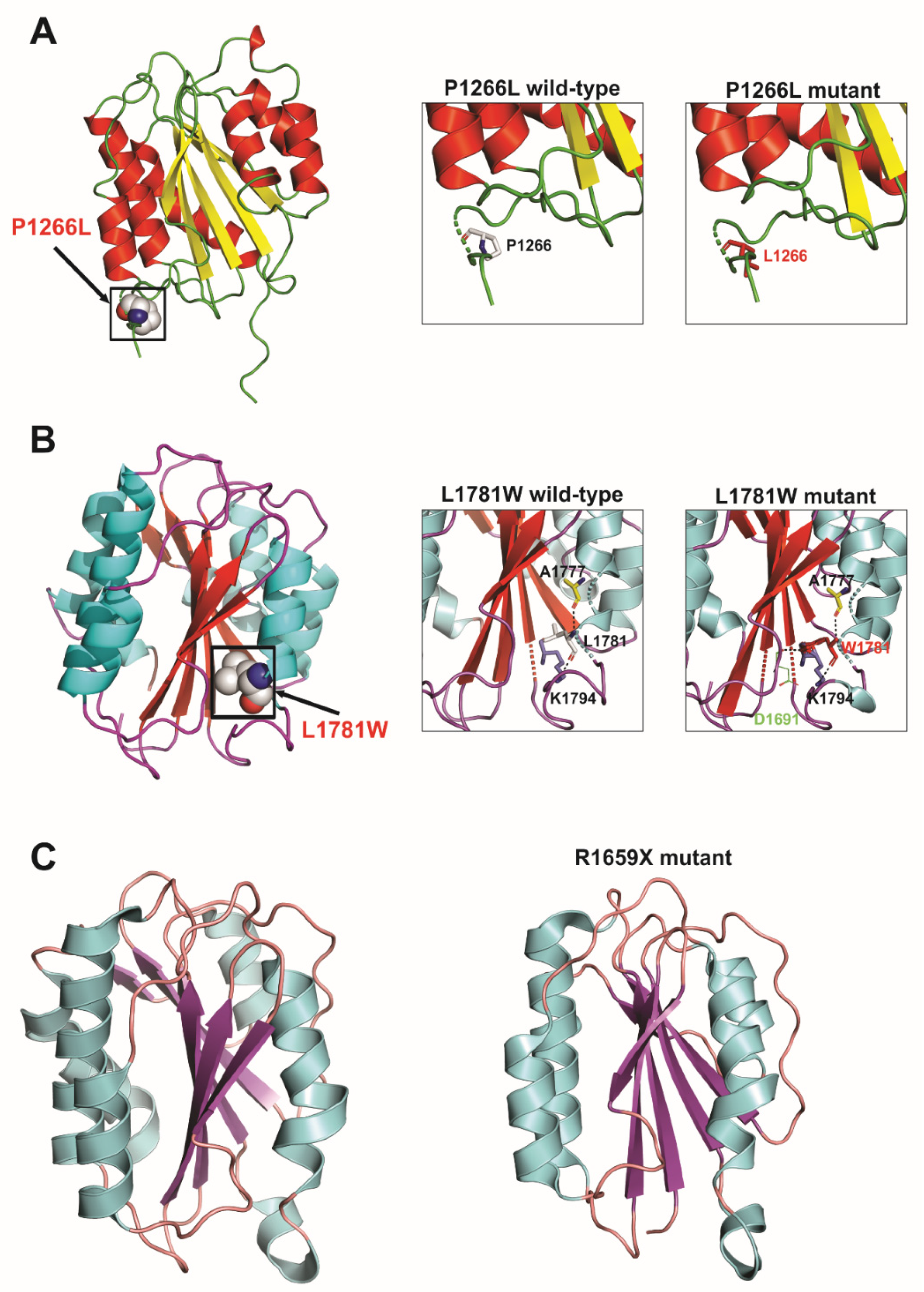
3.1. D protein Structural Analysis
3.2. DNA Methylation Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castaman, G.; Linari, S. Diagnosis and Treatment of von Willebrand Disease and Rare Bleeding Disorders. J. Clin. Med. 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharati, K.P.; Prashanth, U.R. Von Willebrand Disease: An Overview. Indian J. Pharm. Sci. 2011, 73, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodeve, A.; James, P. von Willebrand Disease; Adam, M.P., Ardinger, H.H., Pagon., R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa., G.M., Amemiya., A., Eds.; University of Washington: Seattle, WA, USA, 2011; pp. 365–376. [Google Scholar] [PubMed]
- Orphanet: Von Willebrand Disease. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=903 (accessed on 10 May 2022).
- Şap, F.; Kavakli, T.; Kavakli, K.; Dizdarer, C. The Prevalence of von Willebrand Disease and Significance of in Vitro Bleeding Time (PFA-100) in von Willebrand Disease Screening in the İzmir Region. Turk. J. Hematol. 2013, 30, 40. [Google Scholar] [CrossRef]
- Borhany, M.; Shamsi, T.; Naz, A.; Farzana, T.; Ansari, S.; Nadeem, M.; Rehman, Z.U.; Sangii, Z. Clinical features and types of von Willebrand disease in Karachi. Clin. Appl. Thromb. 2010, 17, E102–E105. [Google Scholar] [CrossRef] [PubMed]
- Mancum, D.J.; Tuley, E.A.; Westfield, L.A.; Worrall, N.K.; Shelton-Inloes, B.B.; Sorace, J.M.; Alevy, Y.G.; Sadler, J.E. The Journal of Biological Chemistry Structure of the Gene for Human von Willebrand Factor*. J. Biol. Chem. 1989, 264, 19514–19527. [Google Scholar] [CrossRef]
- Ahmed, S.; Naz, A.; Yadegari, H.; Driesen, J.; Shamsi, T.; Ahmed, N.; Tariq, S.; Amanat, S.T.; Razik, F.-E.; Nadeem, M.; et al. Molecular Genetics Analysis of Von Williebrand Disease: Studies in Cohort Pakistani Patients. Blood 2015, 126, 4689. [Google Scholar] [CrossRef]
- Kasatkar, P.; Shetty, S.; Ghosh, K. Genetic Heterogeneity in a Large Cohort of Indian Type 3 von Willebrand Disease Patients. PLoS ONE 2014, 9, e92575. [Google Scholar] [CrossRef]
- Sutherland, M.S.; Keeney, S.; Bolton-maggs, P.H.B.; Hay, C.R.M.; Will, A.; Cumming, A.M. The mutation spectrum associated with type 3 von Willebrand disease in a cohort of patients from the north west of England. Haemophilia 2009, 15, 1048–1057. [Google Scholar] [CrossRef]
- Mandava, M.; Lazarchick, J.; Curl, E.; Bergmann, S. A Unique Case of Type 3 Von Willebrand Disease. Blood 2018, 132, 5031. [Google Scholar] [CrossRef]
- Chraszczewska, M.; Sokół, K.; Kuczkiewicz-Siemion, O.; Prochorec-Sobieszek, M.; Szumera-Ciećkiewicz, A. Acquired von Willebrand syndrome associated with plasma cell myeloma—A rare histopathological bone marrow image. Pol. J. Pathol. 2020, 70, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Sottilotta, G.; Luise, F.; Massara, E.; Oriana, V.; Piromalli, A. Efficacy of Octocog Alfa (Advate) in a Child with Type 3 von Willebrand Disease and Alloantibodies. J. Clin. Med. 2017, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, P.D.; Connell, N.T.; Ameer, B.; Di Paola, J.; Eikenboom, J.; Giraud, N.; Haberichter, S.; Jacobs-Pratt, V.; Konkle, B.; McLintock, C.; et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021, 5, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.L.; Hultin, M.B.; James, A.H.; Manco-Johnson, M.J.; Montgomery, R.R.; Ortel, T.L.; Rick, M.E.; Sadler, J.E.; Weinstein, M.; Yawn, B.P. von Willebrand disease (VWD): Evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) expert panel report (USA). Haemophilia 2008, 14, 171–232. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J. Classification of von Willebrand disease in the context of modern contemporary von Willebrand factor testing methodologies. Res. Pract. Thromb. Haemost. 2020, 4, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.; Mundell, G.; Grabell, J.; Hopman, W.M.; Rapson, D.; Lillicrap, D.; James, P. Generation and validation of the Condensed MCMDM-1VWD Bleeding Questionnaire for von Willebrand disease. J. Thromb. Haemost. 2008, 6, 2062–2066. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Purification of nucleic acids by extraction with phenol: Chloroform. Cold Spring Harb. Protoc. 2006, 2006, pdb.prot4455. [Google Scholar] [CrossRef]
- Merriman, B.; Torrent, I.; Rothberg, J.M. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis 2012, 33, 3397–3417. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Amar, A.; Majmundar, A.J.; Ullah, I.; Afzal, A.; Braun, D.A.; Shril, S.; Daga, A.; Jobst-Schwan, T.; Ahmad, M.; Sayer, J.A.; et al. Gene panel sequencing identifies a likely monogenic cause in 7% of 235 Pakistani families with nephrolithiasis. Hum. Genet. 2019, 138, 211. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. Mutation Taster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Mohl, A.; Marschalek, R.; Masszi, T.; Nagy, E.; Obser, T.; Oyen, F.; Sallai, K.; Bodó, I.; Schneppenheim, R. An Alu-mediated novel large deletion is the most frequent cause of type 3 von Willebrand disease in Hungary. J. Thromb. Haemost. 2008, 6, 1729–1735. [Google Scholar] [CrossRef]
- Bowman, M.; Tuttle, A.; Notley, C.; Brown, C.; Tinlin, S.; Deforest, M.; Leggo, J.; Blanchette, V.; Lillicrap, D.; James, P.; et al. The genetics of Canadian type 3 von Willebrand disease: Further evidence for co-dominant inheritance of mutant alleles. J. Thromb. Haemost. 2013, 11, 512–520. [Google Scholar] [CrossRef]
- Ahmed, S.; Yadegari, H.; Naz, A.; Biswas, A.; Budde, U.; Saqlain, N.; Amanat, S.; Tariq, S.; Raziq, F.; Masood, S.; et al. Characterization of the mutation spectrum in a Pakistani cohort of type 3 von Willebrand disease. Haemophilia 2019, 25, 1035–1044. [Google Scholar] [CrossRef]
- Borràs, N.; Batlle, J.; Pérez-Rodríguez, A.; López-Fernández, M.F.; Rodríguez-Trillo, Á.; Lourés, E.; Cid, A.R.; Bonanad, S.; Cabrera, N.; Moret, A.; et al. Molecular and clinical profile of von Willebrand disease in Spain (PCM-EVW-ES): Comprehensive genetic analysis by next-generation sequencing of 480 patients. Haematologica 2017, 102, 2005. [Google Scholar] [CrossRef]
- James, P.D.; Goodeve, A.C. von Willebrand Disease. Genet. Med. 2011, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Sadler, J.E.; Budde, U.; Eikenboom, J.C.; Favaloro, E.J.; Hill, F.G.; Holmberg, L.; Ingerslev, J.; Lee, C.A.; Lillicrap, D.; Mannucci, P.M.; et al. Update on the pathophysiology and classification of von Willebrand disease: A report of the Subcommittee on von Willebrand Factor. J. Thromb. Haemost. 2006, 4, 2103–2114. [Google Scholar] [CrossRef]
- Baronciani, L.; Cozzi, G.; Canciani, M.T.; Peyvandi, F.; Srivastava, A.; Federici, A.B.; Mannucci, P.M. Molecular defects in type 3 von Willebrand disease: Updated results from 40 multiethnic patients. Blood Cells Mol. Dis. 2003, 30, 264–270. [Google Scholar] [CrossRef]
- Kuldanek, S.A.; Venkataraman, S.; Randall, W.; Balakrishnan, I.; Vibhakar, R.; di Paola, J.A. Epigenetic Regulation of Von Willebrand Factor Gene Expression May Contribute to Von Willebrand Disease Severity. Blood 2014, 124, 470. [Google Scholar] [CrossRef]
- Connell, N.T.; Flood, V.H.; Brignardello-Petersen, R.; Abdul-Kadir, R.; Arapshian, A.; Couper, S.; Grow, J.M.; Kouides, P.; Laffan, M.; Lavin, M.; et al. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv. 2021, 5, 301–325. [Google Scholar] [CrossRef]
- Yadegari, H.; Biswas, A.; Ahmed, S.; Naz, A.; Oldenburg, J. von Willebrand factor propeptide missense variants affect anterograde transport to Golgi resulting in ER retention. Hum. Mutat. 2021, 42, 731–744. [Google Scholar] [CrossRef]
- Purvis, A.R.; Gross, J.; Dang, L.T.; Huang, R.-H.; Kapadia, M.; Townsend, R.R.; Sadler, J.E. Two Cys residues essential for von Willebrand factor multimer assembly in the Golgi. Proc. Natl. Acad. Sci. USA 2007, 104, 15647–15652. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Leksa, N.C.; Chhabra, E.S.; Arndt, J.W.; Lu, Q.; Knockenhauer, K.E.; Peters, R.T.; Springer, T.A. The von Willebrand factor D′D3 assembly and structural principles for factor VIII binding and concatemer biogenesis. Blood 2019, 133, 1523–1533. [Google Scholar] [CrossRef]
- Hassan, M.I.; Saxena, A.; Ahmad, F. Structure and function of von Willebrand factor: The protein that is deficient and/or abnormal in inherited von Willebrand disease. Blood Coagul. Fibrinolysis 2012, 23, 11–22. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
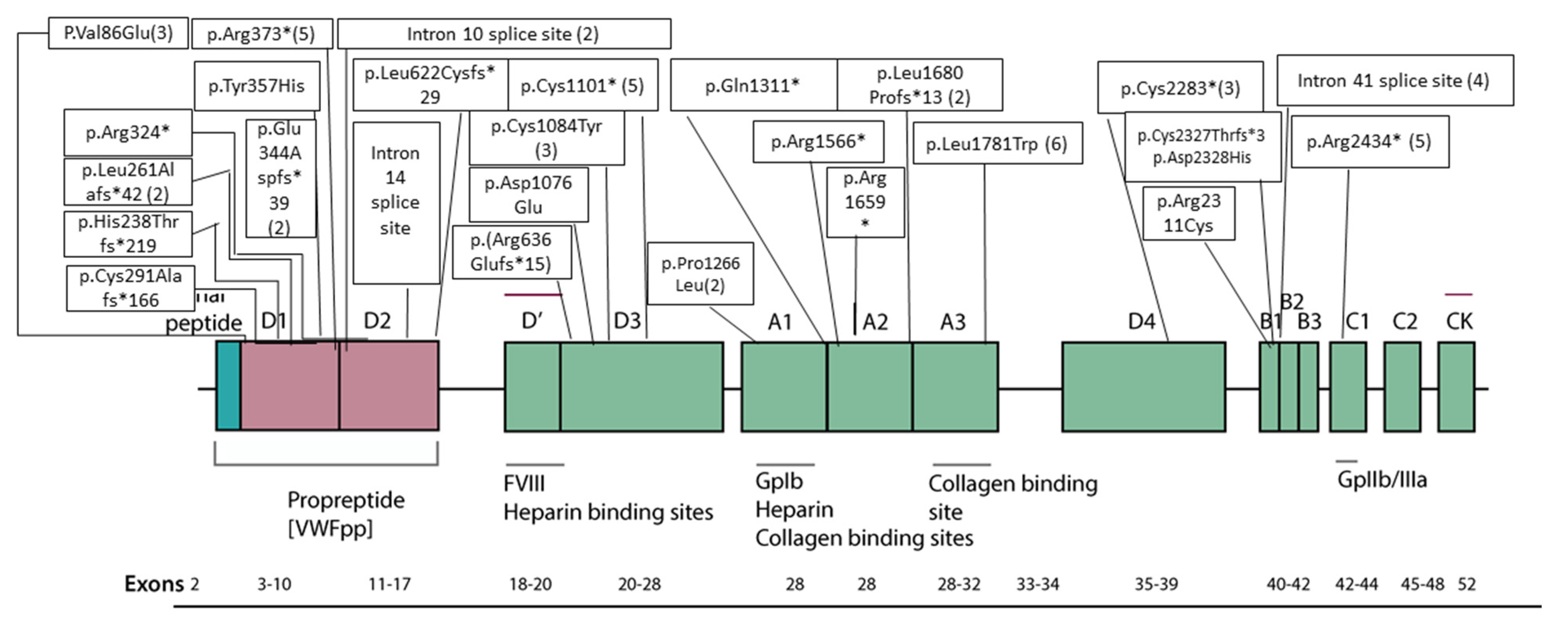
| Family- Individual | Age (Years) | Sex | VWF Level IU/dL | Bleeding Score | Nucleotide Change | Protein Change | Type of Mutation | Exon/Intron and Zygosity | In Silico Severity Scores | Evolutionary Conservation | gnomAD (H/h/T) | rs ID | Reported/ Novel |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| vWD01 | 30 | F | 1.5 | 19 | g.6180502G>A c.1117C>T | p.Arg373* | Nonsense | Ex.10/Hom | NA | NA | 0/6/251032 (All) 0/4/30550(SA) | rs62643625 | Reported |
| vWD02 | 29 | F | 1.1 | 23 | g.6180502G>A c.1117C>T | p.Arg373* | Nonsense | Ex.10/Hom | NA | NA | 0/6/251032 (All) 0/4/30550(SA) | rs62643625 | Reported |
| vWD03 | 9 | F | 1.2 | 21 | g.6091158C>T c.7082-1G>A | NA | Splice site | In. 41/Hom | Splice score—100% | NA | NR | NA | Novel (This study) |
| vWD04 | 17 | F | 0.9 | 24 | g.6091158C>T c.7082-1G>A | NA | Splice site | In. 41/Hom | Splice score—100% | NA | NR | NA | Novel (This study) |
| vWD05 | 29 | F | 1.5 | 15 | g.6166105delG c.1864delC | p.Leu622Cysfs*29 | Frameshift | Ex. 15/Hom | NA | NA | NR | NA | Novel (This study) |
| vWD6 | 14 | F | 2.2 | 19 | g.6094781G>T c.6849C>A | p.Cys2283* | Nonsense | Ex.39/Hom | NA | NA | NR | NA | Novel (This study) |
| vWD7 | 7 | M | 1.7 | 16 | g.6094781G>T c.6849C>A | p.Cys2283* | Nonsense | Ex.39/Hom | NA | NA | NR | NA | Novel (This study) |
| vWD8 | 7 | F | 1.4 | 12 | g.6127545delA c.5039delT | p.Leu1680Profs*13 | Frameshift | Ex. 28/Hom | NA | NA | NR | NA | Novel (This study) |
| vWD9 | 7 | M | 1.5 | 23 | g.6132873G>T c.3303C>A | p.Cys1101* | Nonsense | Ex.25/Hom | NA | NA | NR | NA | Bowman (2013) J Thromb Haemost 11, 512 |
| vWD10 | 21 | F | 1.4 | 21 | g.6128653G>A c.3931C>T | p.Gln1311* | Nonsense | Ex.28/Hom | NA | NA | 0/13/250944 (All) 0/12/30604 (SA) | rs267607337 | Casana (2000) Br J Haematol 111, 552 and others |
| vWD11 | 18 | M | 1.9 | 15 | g.6091158C>T c.7082-1G>A | NA | Splice site | In. 41/Hom | Splice score—100% | NA | NR | NA | Novel (This study) |
| vWD14 | 26 | M | 3.2 | 19 | g.6180462C>T c.1156+1G>A | NA | Splice site | In. 10/Het | Splice score—100% | NA | NR | NA | Novel (This study) |
| vWD15 | 24 | M | 2.1 | 13 | g.6180462C>T c.1156+1G>A | NA | Splice site | In. 10/Het | Splice score—100% | NA | NR | NA | Novel (This study) |
| vWD18 | 8 | M | 0.3 | 18 | g.6132873G>T c.3303C>A | p.Cys1101* | Nonsense | Ex.25/Hom | NA | NA | NR | NA | Bowman (2013) J Thromb Haemost 11, 512 |
| vWD19 | 35 | M | 1.7 | 15 | g.6127888G>A c.4696C>T | p.Arg1566* | Nonsense | Ex.28/Hom | NA | NA | 0/2/125338 (All) | rs61750112 | Yes (Indian + Chinese studies) |
| vWD20 | 11 | F | 1.9 | 9 | g.6220098A>T c.257T>A | p.Val86Glu | Missense | Ex. 4/Hom | PPh2 = D SIFT = DL MT = DC | Gallus gallus (G. gallus) | NR | NA | Ahmed, 2019 |
| vWD21 | 7 | M | 2.8 | 8 | g.6125368A>C c.5342T>G | p.Leu1781Trp | Missense | Ex. 31/Hom | PPh2 = PD SIFT = DL MT = P | Homo sapiens (H. sapiens) | NR | NA | Ahmed, 2019 |
| vWD22 | 26 | F | 0.3 | 11 | g.6125368A>C c.5342T>G | p.Leu1781Trp | Missense | Ex. 31/Hom | PPh2 = PD SIFT = DL MT = P | H. sapiens | NR | NA | Ahmed, 2019 |
| vWD23 | 17 | M | 3.2 | 12 | g.6125368A>C c.5342T>G | p.Leu1781Trp | Missense | Ex. 31/Hom | PPh2 = PD SIFT = DL MT = P | H. sapiens | NR | NA | Ahmed, 2019 |
| vWD25 | 20 | M | 1.6 | 17 | g.6132873G>T c.3303C>A | p.Cys1101* | Nonsense | Ex.25/Hom | NA | NA | NR | NA | Bowman (2013) J Thromb Haemost 11, 512 |
| vWD26 | 19 | F | 1 | 19 | g.6132873G>T c.3303C>A | p.Cys1101* | Nonsense | Ex.25/Hom | NA | NA | NR | NA | Bowman (2013) J Thromb Haemost 11, 512 |
| vWD27 | 17 | M | 1.2 | 11 | g.6127609G>A c.4975C>T | p.Arg1659* | Nonsense | Ex.28/Het | NA | NA | 0/16/281716 (All) 0/0/30562 (SA) | rs61750595 | Zhang (1992) Hum Mol Genet 1, 61 and others |
| g.6184595insC c.780InsG | p.Leu261Alafs*42 | Frameshift | Ex. 7/Het | NA | NA | NR | rs760130928 | Novel (This study) | |||||
| vWD28 | 19 | M | 3.4 | 14 | g.6091158C>T c.7082-1G>A | NA | Splice site | In. 41/Hom | Splice score—100% | NA | NR | NA | Novel (This study) |
| vWD29 | 20 | F | 1.9 | 13 | g.6166066delC c.1905delC | p.Arg636Glufs*15 | Frameshift | Ex. 15/Het | NA | NA | NR | NA | Novel (This study) |
| vWD30 | 13 | M | 3.1 | 15 | g.6085414G>A c.7300C>T | p.Arg2434* | Nonsense | Ex. 43/Hom | NA | NA | 0/2/31380 (All) NR (SA) | rs62643640 | Baronciani (2003) Blood Cells Mol Dis 30, 264 and others |
| vWD32 | 30 | F | 1.5 | 8 | g.6125368A>C c.5342T>G | p.Leu1781Trp | Missense | Ex. 31/Hom | PPh2 = PD SIFT = DL MT = P | H. sapiens | NR | NA | Ahmed, 2019 |
| vWD33 | 29 | M | 4.5 | 14 | g.6220098A>T c.257T>A | p.Val86Glu | Missense | Ex. 4/Hom | PPh2 = D SIFT = DL MT = DC | G. gallus | NR | NA | Ahmed, 2019 |
| vWD35 | 35 | M | 4.5 | 24 | g.6085414G>A c.7300C>T | p.Arg2434* | Nonsense | Ex. 43/Hom | NA | NA | 0/2/31380 (All) NR (SA) | rs62643640 | Baronciani (2003) Blood Cells Mol Dis 30, 264 and others |
| vWD36 | 34 | F | 2.8 | 20 | g.6180502G>A c.1117C>T | p.Arg373* | Nonsense | Ex.10/Hom | NA | NA | 0/6/251032 (All) 0/4/30550(SA) | rs62643625 | Baronciani (2000) Thromb Haemost 84, 536 |
| vWD38 | 11 | F | 2.1 | 15 | g.6125368A>C c.5342T>G | p.Leu1781Trp | Missense | Ex. 31/Hom | PPh2 = PD SIFT = DL MT = P | H. sapiens | NR | NA | Ahmed, 2019 |
| vWD39 | 17 | M | 1.8 | 14 | g.6132925C>T c.3251G>A | p.Cys1084Tyr | Missense | Ex. 25/Hom | PPh2 = D SIFT = DL MT = DC | Danio rerio (D. rerio) | 0/2/124535 (All) 0/2/30464 (SA) | rs759805079 | Novel (This study) |
| vWD40 | 7 | M | 0.9 | 18 | g.6184664delG c.712delG | p.His238Thrfs*219 | Frameshift | Ex. 7/Het | NA | NA | NR | NA | Novel (This study) |
| vWD41 | 40 | F | 1.3 | 23 | g.6085414G>A c.7300C>T | p.Arg2434* | Nonsense | Ex. 43/Hom | NA | NA | 0/2/31380 (All) NR (SA) | rs62643640 | Baronciani (2003) Blood Cells Mol Dis 30, 264 and others |
| vWD42 | 19 | M | 1.4 | 12 | g.6220098A>T c.257T>A | p.Val86Glu | Missense | Ex. 4/Hom | PPh2 = D SIFT = DL MT = DC | G. gallus | NR | NA | Ahmed, 2019 |
| vWD43 | 22 | M | 1.2 | 13 | g.6184504delA c.871delT | p.Cys291Alafs*166 | Frameshift | Ex. 7/Hom | NA | NA | NR | rs1565853817 | Novel (This study) |
| vWD44 | 26 | M | 2.9 | 14 | g.6094256G>A c.6931C>T | p.Arg2311Cys | Missense | Ex. 40/Hom | PPh2 = D SIFT = DL MT = DC | Xenopus tropicalis (X. tropicalis) | 0/22/282886 (All) 0/1/30616 (SA) | rs150725355 | Novel (This study) |
| vWD47 | 25 | M | 1.3 | 14 | g.6132925C>T c.3251G>A | p.Cys1084Tyr | Missense | Ex. 25/Hom | PPh2 = D SIFT = DL MT = DC | D. rerio | 0/2/124535 (All) 0/2/30464 (SA) | rs759805079 | Novel (This study) |
| vWD48 | 26 | M | 2.7 | 11 | g.6127545delA c.5039delT | p.Leu1680Profs*13 | Frameshift | Ex. 28/Hom | NA | NA | NR | NA | Novel (This study) |
| vWD49 | 21 | M | 0.3 | 19 | g.6085414G>A c.7300C>T | p.Arg2434* | Nonsense | Ex. 43/Hom | NA | NA | 0/2/31380 (All) NR (SA) | rs62643640 | Baronciani (2003) Blood Cells Mol Dis 30, 264 and others |
| vWD50 | 14 | M | 0.4 | 16 | g.6180502G>A c.1117C>T | p.Arg373* | Nonsense | Ex.10/Hom | NA | NA | 0/6/251032 (All) 0/4/30550(SA) | rs62643625 | Baronciani (2000) Thromb Haemost 84, 536 |
| vWD51 | 18 | F | 1.4 | 23 | g.6132873G>T c.3303C>A | p.Cys1101* | Nonsense | Ex.25/Hom | NA | NA | NR | NA | Bowman (2013) J Thromb Haemost 11, 512 |
| vWD53 | 21 | F | 1.1 | 21 | g.6085414G>A c.7300C>T | p.Arg2434* | Nonsense | Ex. 43/Hom | NA | NA | 0/2/31380 (All) NR (SA) | rs62643640 | Baronciani (2003) Blood Cells Mol Dis 30, 264 and others |
| vWD54 | 23 | F | 2.2 | 17 | g.6127609G>A c.4975C>T | p.Arg1659* | Nonsense | Ex.28/Het | NA | NA | 0/16/281716 (All) 0/0/30562 (SA) | rs61750595 | Zhang (1992) Hum Mol Genet 1, 61 and others |
| vWD55 | 27 | M | 1.5 | 17 | g.6182812G>A c.970C>T | p.Arg324* | Nonsense | Ex.8/Hom | NA | NA | 0/3/251370 (All) 0/2/30616 (SA) | rs61754000 | Schneppenheim (1994) Hum Genet 94, 640 and others |
| vWD56 | 29 | F | 2.3 | 11 | g.6092417_6092426del c.6977-6_6980del | p.Cys2327Thrfs*3 | Frameshift & splice site | Ex. 41/Hom | Splice score—100% | NA | NR | NA | Novel (This study) |
| g.6092415C>G c.6982G>C | p.Asp2328His | Missense | Ex. 41/Hom | PPh2 = D SIFT = DL MT = DC | Ciona intestinalis (C. intestinalis) | NR | NA | Novel (This study) | |||||
| vWD57 | 15 | F | 1.5 | 9 | g.6132925C>T c.3251G>A | p.Cys1084Tyr | Missense | Ex. 25/Hom | PPh2 = D SIFT = DL MT = DC | D. rerio | 0/2/124535 (All) 0/2/30464 (SA) | rs759805079 | Novel (This study) |
| vWD58 | 32 | F | 1.4 | 22 | g.6180502G>A c.1117C>T | p.Arg373* | Nonsense | Ex.10/Hom | NA | NA | 0/6/251032 (All) 0/4/30550(SA) | rs62643625 | Baronciani (2000) Thromb Haemost 84, 536 |
| vWD61 | 22 | F | 2.4 | 19 | g.6166240T>G c.1730-2A>C | NA | Splice site | In. 14/Hom | Splice score—100% | NA | 0/1/133334 (All) 0/1/21944 (SA) | rs61754014 | Novel (This study) |
| vWD62 | 5 | M | 2.3 | 19 | g.6094781G>T c.6849C>A | p.Cys2283* | Nonsense | Ex.39/Hom | NA | NA | NR | NA | Novel (This study) |
| vWD63 | 25 | F | 2.2 | 19 | g.6125368A>C c.5342T>G | p.Leu1781Trp | Missense | Ex. 31/Hom | PPh2 = PD SIFT = DL MT = P | H. sapiens | NR | NA | Ahmed, 2019 |
| vWD66 | 9 | M | 1.3 | 13 | g.6128787G>A c.3797C>T | p.Pro1266Leu | Missense | Ex. 28/Hom | PPh2 = D SIFT = T MT = DC | Mus musculus (M. musculus) | 1/234/281180 (All) 0/7/30468 (SA) | rs61749370 | Holmberg (1993) J Clin Invest 91, 2169 |
| vWD67 | 13 | F | 1.2 | 17 | g.6181537A>G c.1069T>C | p.Tyr357His | Missense | Ex. 9/Het | PPh2 = D SIFT = DL MT = DC | D. rerio | NR | NA | Novel (This study) |
| g.6184594_6184595insC c.780_781insG | p.Leu261Alafs*42 | Frameshift | Ex. 7/Het | NA | NA | 0/2/251064 (All) 0/0/30612 (SA) | NA | Novel (This study) | |||||
| vWD68 | 12 | F | 1.3 | 15 | g.6132948G>C c.3228C>G | p.Asp1076Glu | Missense | Ex. 25/Het | PPh2 = PD SIFT = T MT = DC | M. musculus | NR | NA | Novel (This study) |
| vWD70 | 9 | F | 1.7 | 12 | g.6128787G>A c.3797C>T | p.Pro1266Leu | Missense | Ex. 28/Hom | PPh2 = D SIFT = T MT = DC | M. musculus | 1/234/281180 (All) 0/7/30468 (SA) | rs61749370 | Holmberg (1993) J Clin Invest 91, 2169 |
| vWD71 | 20 | F | 1.8 | 17 | g:6181574_6181575insG c.1031_1032insC | p.Glu344Aspfs*39 | Frameshift | Ex. 9/Het | NA | NA | NR | NA | Novel (This study) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naveed, M.A.; Abid, A.; Ali, N.; Hassan, Y.; Amar, A.; Javed, A.; Qamar, K.; Mustafa, G.; Raza, A.; Saleem, U.; et al. Genetic Alterations, DNA Methylation, Alloantibodies and Phenotypic Heterogeneity in Type III von Willebrand Disease. Genes 2022, 13, 971. https://doi.org/10.3390/genes13060971
Naveed MA, Abid A, Ali N, Hassan Y, Amar A, Javed A, Qamar K, Mustafa G, Raza A, Saleem U, et al. Genetic Alterations, DNA Methylation, Alloantibodies and Phenotypic Heterogeneity in Type III von Willebrand Disease. Genes. 2022; 13(6):971. https://doi.org/10.3390/genes13060971
Chicago/Turabian StyleNaveed, Muhammad Asif, Aiysha Abid, Nadir Ali, Yaqoob Hassan, Ali Amar, Aymen Javed, Khansa Qamar, Ghulam Mustafa, Ali Raza, Umera Saleem, and et al. 2022. "Genetic Alterations, DNA Methylation, Alloantibodies and Phenotypic Heterogeneity in Type III von Willebrand Disease" Genes 13, no. 6: 971. https://doi.org/10.3390/genes13060971