
Potential Composite Digenic Contribution of NPC1 and NOD2 Leading to Atypical Lethal Niemann-Pick Type C with Initial Crohn’s Disease-like Presentation: Genotype-Phenotype Correlation Study

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Sample Collection and Sequencing
2.3. Simulation Analysis
3. Results
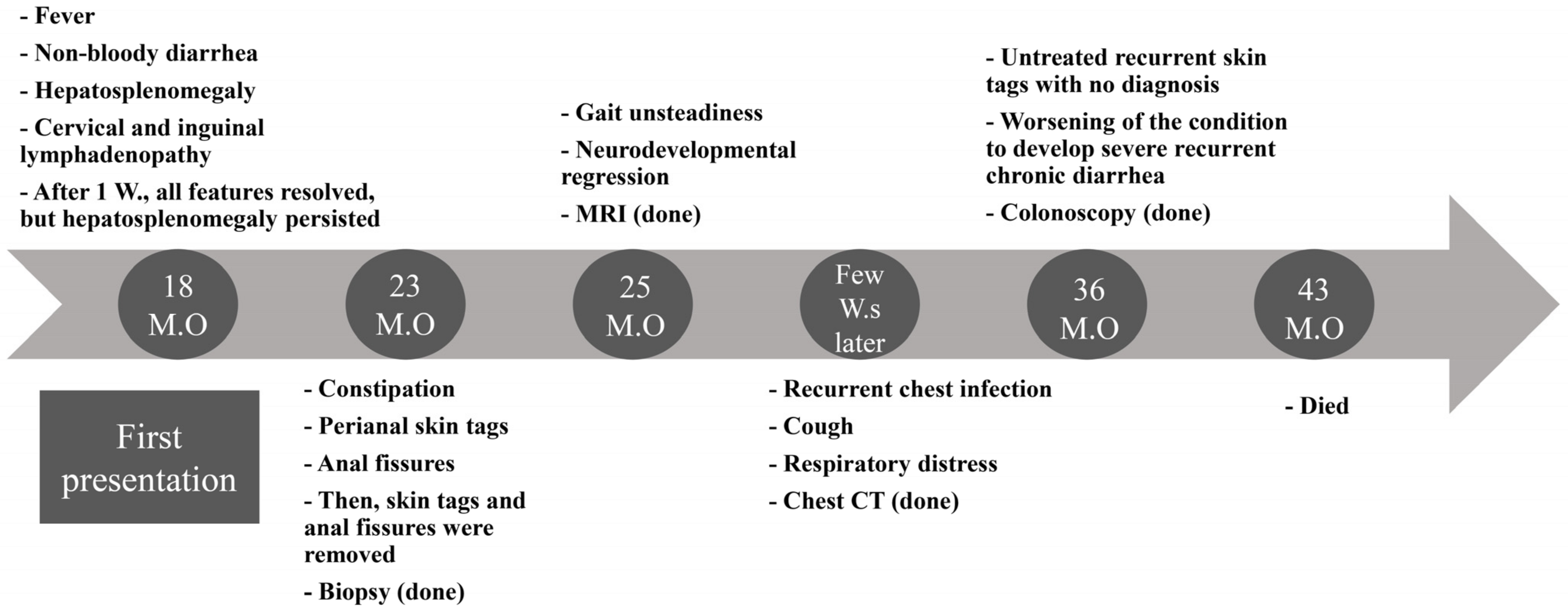
3.1. Overview of the Clinical Presentation
3.2. Initial Presentation
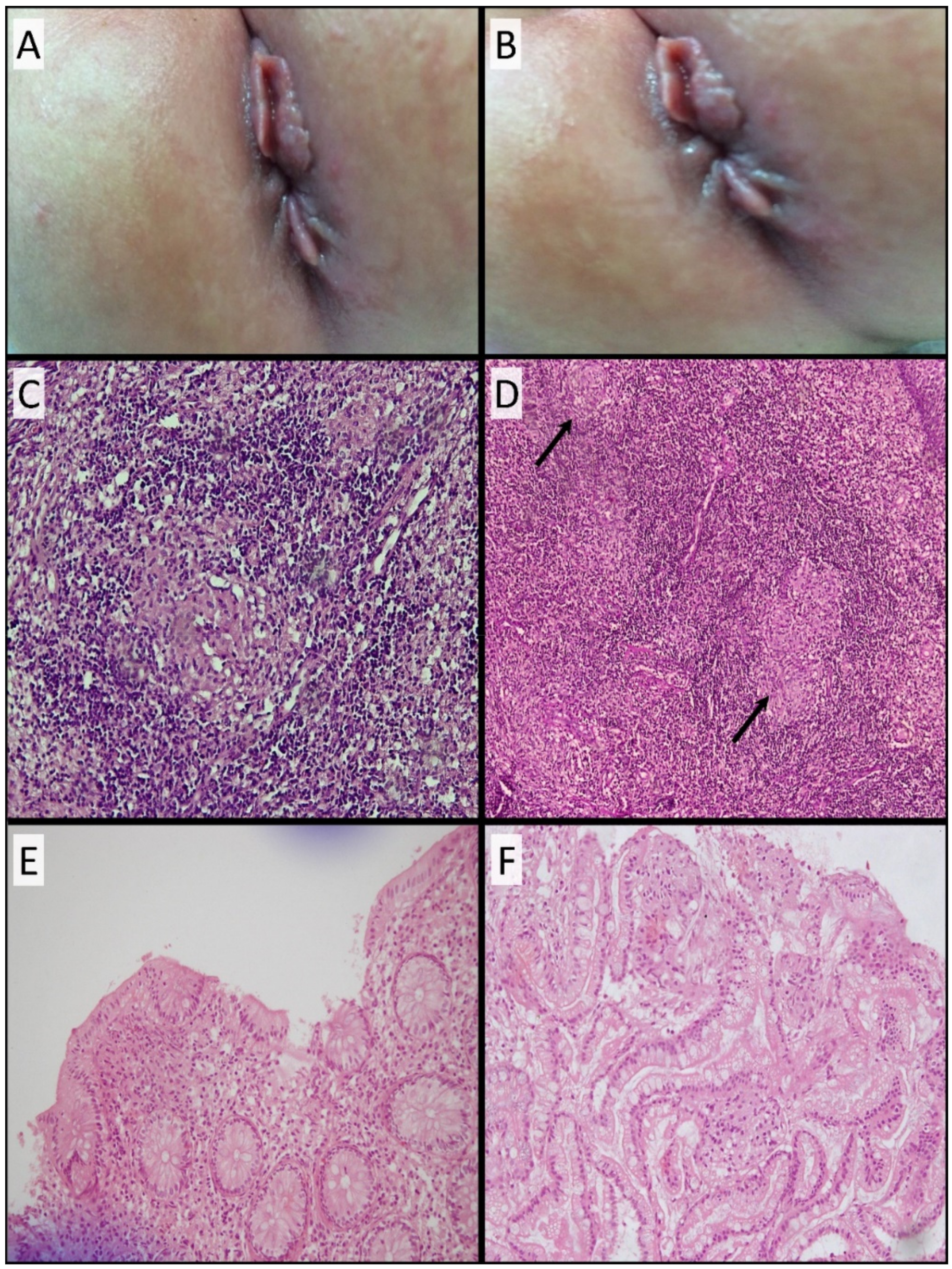
3.3. Development of Skin Tags and Anal Fissures and Histopathological Findings
3.4. Development of Neurological Manifestations and MRI Evaluation
3.5. Development of Respiratory Symptoms and Subsequent Neurological Deterioration
3.6. Recurrent Chest Infections and Chest CT Findings
3.7. Colonoscopy and Histopathological Findings
3.8. Treatment Regimen
3.9. Dilemma of Establishing Definitive Differential Clinical Diagnosis
3.10. Genetic Findings
3.11. Simulation Analysis of NOD2 and NPC1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kolodny, E.H. Niemann–Pick Disease. Curr Opin Hematol. 2000, 7, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Jones, S.A.; Soran, H.; Diaz, G.A.; Lippa, N.; Thurberg, B.L.; Culm-Merdek, K.; Shamiyeh, E.; Inguilizian, H.; Cox, G.F.; et al. Successful within-patient dose escalation of olipudase alfa in acid sphingomyelinase deficiency. Mol. Genet. Metab. 2015, 116, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef]
- Patterson, M.C.; Mengel, E.; Wijburg, F.A.; Muller, A.; Schwierin, B.; Drevon, H.; Vanier, M.T.; Pineda, M. Disease and patient characteristics in NP-C patients: Findings from an international disease registry. Orphanet J. Rare Dis. 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walterfang, M.; Chien, Y.H.; Imrie, J.; Rushton, D.; Schubiger, D.; Patterson, M.C. Dysphagia as a risk factor for mortality in Niemann-Pick disease type C: Systematic literature review and evidence from studies with miglustat. Orphanet J. Rare Dis. 2012, 7, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlig, H.H.; Schwerd, T. From Genes to Mechanisms: The Expanding Spectrum of Monogenic Disorders Associated with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 202–212. [Google Scholar] [CrossRef]
- Caso, F.; Galozzi, P.; Costa, L.; Sfriso, P.; Cantarini, L.; Punzi, L. Review: Autoinflammatory granulomatous diseases: From Blau syndrome and early-onset sarcoidosis to NOD2-mediated disease and Crohn’s disease. RMD Open 2015, 1, e000097. [Google Scholar] [CrossRef]
- Yao, Q.; Li, E.; Shen, B. Autoinflammatory disease with focus on NOD2-associated disease in the era of genomic medicine. Autoimmunity 2019, 52, 48–56. [Google Scholar] [CrossRef]
- Macías-Vidal, J.; Girós, M.; Guerrero, M.; Gascón, P.; Serratosa, J.; Bachs, O.; Coll, A.J. The proteasome inhibitor bortezomib reduced cholesterol accumulation in fibroblasts from Niemann-Pick type C patients carrying missense mutations. FEBS J. 2014, 281, 4450–4466. [Google Scholar] [CrossRef]
- Wraith, J.E.; Sedel, F.; Pineda, M.; Wijburg, F.A.; Hendriksz, C.J.; Fahey, M.; Walterfang, M.; Patterson, M.C.; Chadha-Boreham, H.; Kolb, S.A. Niemann-Pick type C Suspicion Index tool: Analyses by age and association of manifestations. J. Inherit. Metab. Dis. 2014, 37, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwerd, T.; Pandey, S.; Yang, H.-T.; Bagola, K.; Jameson, E.; Jung, J.; Lachmann, R.H.; Shah, N.; Patel, S.Y.; Booth, C.; et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann-Pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn’s disease. Gut 2017, 66, 1060–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.T.; Williams, W.J. Granulomatous inflammation—A review. J. Clin. Pathol. 1983, 36, 723. [Google Scholar] [CrossRef] [PubMed]
- Pagán, A.J.; Ramakrishnan, L. The Formation and Function of Granulomas. Annu. Rev. Immunol. 2018, 36, 639–665. [Google Scholar] [CrossRef]
- Shah, K.K.; Pritt, B.S.; Alexander, M.P. Histopathologic review of granulomatous inflammation. J. Clin. Tuberc. Other Mycobact. Dis. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Azab, B.; Dardas, Z.; Rabab’h, O.; Srour, L.; Telfah, H.; Hatmal, M.M.; Mustafa, L.; Rashdan, L.; Altamimi, E. Enteric anendocrinosis attributable to a novel Neurogenin-3 variant. Eur. J. Med. Genet. 2020, 63, 103981. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef]
- Mokhtariye, A.; Hagh-Nazari, L.; Varasteh, A.R.; Keyfi, F. Diagnostic methods for Lysosomal Storage Disease. Rep. Biochem. Mol. Biol. 2019, 7, 119. [Google Scholar]
- Janssen, C.E.I.; Rose, C.D.; De Hertogh, G.; Martin, T.M.; Bader Meunier, B.; Cimaz, R.; Harjacek, M.; Quartier, P.; Ten Cate, R.; Thomee, C.; et al. Morphologic and immunohistochemical characterization of granulomas in the nucleotide oligomerization domain 2-related disorders Blau syndrome and Crohn disease. J. Allergy Clin. Immunol. 2012, 129, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Yadav, D.; Puranik, N.; Guleria, R.; Jin, J.O. Sarcoidosis: Causes, Diagnosis, Clinical Features, and Treatments. J. Clin. Med. 2020, 9, 1081. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, A.P.; Fisher, S.A.; Mirza, M.M.; King, K.; Hampe, J.; Croucher, P.J.P.; Mascheretti, S.; Sanderson, J.; Forbes, A.; Mansfield, J.; et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology 2002, 122, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, A.M.; Ombrello, M.J. Using genes to triangulate the pathophysiology of granulomatous autoinflammatory disease: NOD2, PLCG2 and LACC1. Int. Immunol. 2018, 30, 205–213. [Google Scholar] [CrossRef]
- Saulsbury, F.T.; Wouters, C.H.; Martin, T.M.; Austin, C.R.; Doyle, T.M.; Goodwin, K.A.; Rosé, C.D. Incomplete penetrance of the NOD2 E383K substitution among members of a pediatric granulomatous arthritis pedigree. Arthritis Rheum. 2009, 60, 1804–1806. [Google Scholar] [CrossRef]
- Girardelli, M.; Loganes, C.; Pin, A.; Stacul, E.; Decleva, E.; Vozzi, D.; Baj, G.; De Giacomo, C.; Tommasini, A.; Bianco, A.M. Novel NOD2 Mutation in Early-Onset Inflammatory Bowel Phenotype. Inflamm. Bowel Dis. 2018, 24, 1204–1212. [Google Scholar] [CrossRef]
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nanba, E.; Ninomiya, H.; Higaki, K.; Taniguchi, M.; Zhang, H.; Akaboshi, S.; Watanabe, Y.; Takeshima, T.; Inui, K.; et al. NPC1 gene mutations in Japanese patients with Niemann-Pick disease type C. Hum. Genet. 1999, 105, 10–16. [Google Scholar] [CrossRef]
- Macías-Vidal, J.; Gort, L.; Lluch, M.; Pineda, M.; Coll, M.J. Nonsense-mediated mRNA decay process in nine alleles of Niemann-Pick type C patients from Spain. Mol. Genet. Metab. 2009, 97, 60–64. [Google Scholar] [CrossRef]
- Macías-Vidal, J.; Rodríguez-Pascau, L.; Sánchez-Ollé, G.; Lluch, M.; Vilageliu, L.; Grinberg, D.; Coll, M. Molecular analysis of 30 Niemann-Pick type C patients from Spain. Clin. Genet. 2011, 80, 39–49. [Google Scholar] [CrossRef]
- Jahnova, H.; Dvorakova, L.; Vlaskova, H.; Hulkova, H.; Poupetova, H.; Hrebicek, M.; Jesina, P. Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: A surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J. Rare Dis. 2014, 9, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Granero, F.; Blanco-Kelly, F.; Sanchez-Jimeno, C.; Avila-Fernandez, A.; Arteche, A.; Bustamante-Aragones, A.; Rodilla, C.; Rodríguez-Pinilla, E.; Riveiro-Alvarez, R.; Tahsin-Swafiri, S.; et al. Comparison of the diagnostic yield of aCGH and genome-wide sequencing across different neurodevelopmental disorders. NPJ Genom. Med. 2021, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Dike, C.R.; Bernat, J.; Bishop, W.; DeGeeter, C. Niemann-Pick disease type C presenting as very early onset inflammatory bowel disease. BMJ Case Rep. 2019, 12, 10–12. [Google Scholar] [CrossRef]
- Jolliffe, D.S.; Sarkany, I. Niemann-Pick type III and Crohn’s disease. J. R. Soc. Med. 1983, 76, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Steven, L.C.; Driver, C.P. Niemann-Pick disease type C and Crohn’s disease. Scott. Med. J. 2005, 50, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Cavounidis, A.; Uhlig, H.H. Crohn’s Disease in Niemann–Pick Disease Type C1: Caught in the Cross-Fire of Host-Microbial Interactions. Dig. Dis. Sci. 2018, 63, 811–813. [Google Scholar] [CrossRef] [Green Version]
- Simpson, W.L.; Mendelson, D.; Wasserstein, M.P.; McGovern, M.M. Imaging Manifestations of Niemann-Pick Disease Type B. Am. J. Roentgenol. 2012, 194, W12–W19. [Google Scholar] [CrossRef]
- Yilmaz, B.S.; Baruteau, J.; Rahim, A.A.; Gissen, P. Clinical and molecular features of early infantile niemann pick type c disease. Int. J. Mol. Sci. 2020, 21, 5059. [Google Scholar] [CrossRef]
- Staretz-Chacham, O.; Aviram, M.; Morag, I.; Goldbart, A.; Hershkovitz, E. Pulmonary involvement in Niemann-Pick C type 1. Eur. J. Pediatr. 2018, 177, 1609–1615. [Google Scholar] [CrossRef]
- Wouters, C.H.; Maes, A.; Foley, K.P.; Bertin, J.; Rose, C.D. Blau Syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr. Rheumatol. Online J. 2014, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Gazzo, A.; Raimondi, D.; Daneels, D.; Moreau, Y.; Smits, G.; Van Dooren, S.; Lenaerts, T. Understanding mutational effects in digenic diseases. Nucleic Acids Res. 2017, 45, e140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46W, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi,psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Spassov, V.Z.; Yan, L. pH-selective mutagenesis of protein–protein interfaces: In silico design of therapeutic antibodies with prolonged half-life. Proteins 2013, 81, 704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spassov, V.Z.; Yan, L. A fast and accurate computational approach to protein ionization. Protein Sci. 2008, 17, 1955–1970. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Qian, H.; Wu, X.; Du, X.; Yao, X.; Zhao, X.; Lee, J.; Yang, H.; Yan, N. Structural Basis of Low-pH-Dependent Lysosomal Cholesterol Egress by NPC1 and NPC2. Cell 2020, 182, 98–111.e18. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Category | Requested Lab Test | Result |
|---|---|---|
| Serology | Pathogen (CMV, HBV, HCV, Parvo virus, EBV, Widal and Brucella, TB, HIV, HSVI, HSVII, flu A, flu B and H1N1) | Negative |
| C3 and C4 | Normal | |
| Rheumatoid factor (RF) | Normal | |
| Antinuclear antibodies (ANA) | Normal | |
| Stool analysis | WBC | Elevated |
| Reducing substances | Positive | |
| Infectious agent | Negative | |
| Metabolic workup | Amino acid analysis and organic acid analysis | Normal |
| Multiplex newborn screening test (Pompe, Fabry, mucopolysaccharidosis type 1 Krabbe, Gaucher and Niemann–Pick disease type A/B) | Normal | |
| Immunology workup | Immunoglobulins levels | Normal |
| Flow cytometry | Elevated CD19 B cells | |
| Burst test | Normal | |
| Miscellaneous tests | LDH | Elevated |
| Ferritin | Elevated | |
| α fetoprotein | Normal | |
| B-HCG | Normal | |
| CK | Normal | |
| Lipid profile | Low HDL |
| Gene | Variant Coordinate | Transcript ID | Exon | Variant Description | In-silico Prediction | |||
|---|---|---|---|---|---|---|---|---|
| dbSNP ID | HGVS cDNA aa | Zygo | Max AF gnomADv3 gnomADv2 | PROVEAN REVEL SIFT (Score) | ||||
| NOD2 | hg38:chr16:50711101hg19:chr16:50745012 | NM_022162.2 | 4/12 | rs150078153 | c.1190C>T p.(Pro397Leu) | HET | V3: 0.0003087 V2: 0.0002711 | Deleterious (−5.88) Benign (0.4399) Damaging (0.000) |
| NPC1 | hg38:chr18:23568933hg19:chr18:21148897 | NM_000271.4 | 4/25 | rs759075595 | c.352_353delAG p.(Gln119ValfsTer8) | HOM | V3:0 V2: 0.00004620 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azab, B.; Rabab’h, O.; Aburizeg, D.; Mohammad, H.; Dardas, Z.; Mustafa, L.; Khasawneh, R.A.; Awad, H.; Hatmal, M.M.; Altamimi, E. Potential Composite Digenic Contribution of NPC1 and NOD2 Leading to Atypical Lethal Niemann-Pick Type C with Initial Crohn’s Disease-like Presentation: Genotype-Phenotype Correlation Study. Genes 2022, 13, 973. https://doi.org/10.3390/genes13060973
Azab B, Rabab’h O, Aburizeg D, Mohammad H, Dardas Z, Mustafa L, Khasawneh RA, Awad H, Hatmal MM, Altamimi E. Potential Composite Digenic Contribution of NPC1 and NOD2 Leading to Atypical Lethal Niemann-Pick Type C with Initial Crohn’s Disease-like Presentation: Genotype-Phenotype Correlation Study. Genes. 2022; 13(6):973. https://doi.org/10.3390/genes13060973
Chicago/Turabian StyleAzab, Bilal, Omar Rabab’h, Dunia Aburizeg, Hashim Mohammad, Zain Dardas, Lina Mustafa, Ruba A. Khasawneh, Heyam Awad, Ma’mon M. Hatmal, and Eyad Altamimi. 2022. "Potential Composite Digenic Contribution of NPC1 and NOD2 Leading to Atypical Lethal Niemann-Pick Type C with Initial Crohn’s Disease-like Presentation: Genotype-Phenotype Correlation Study" Genes 13, no. 6: 973. https://doi.org/10.3390/genes13060973
APA StyleAzab, B., Rabab’h, O., Aburizeg, D., Mohammad, H., Dardas, Z., Mustafa, L., Khasawneh, R. A., Awad, H., Hatmal, M. M., & Altamimi, E. (2022). Potential Composite Digenic Contribution of NPC1 and NOD2 Leading to Atypical Lethal Niemann-Pick Type C with Initial Crohn’s Disease-like Presentation: Genotype-Phenotype Correlation Study. Genes, 13(6), 973. https://doi.org/10.3390/genes13060973