
SINEs as Credible Signs to Prove Common Ancestry in the Tree of Life: A Brief Review of Pioneering Case Studies in Retroposon Systematics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Actual Outcomes of the SINE Method
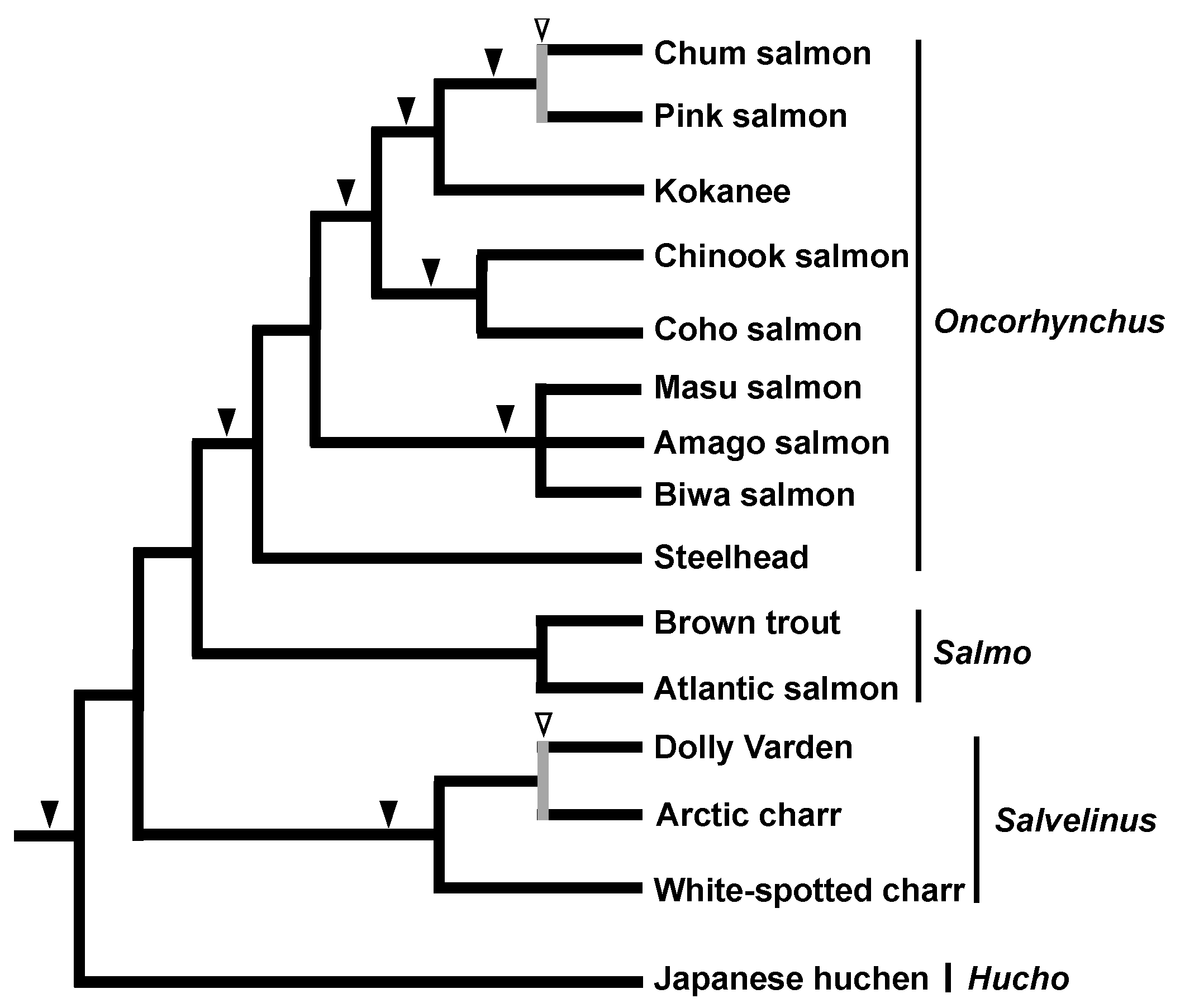
2.1. Salmonid Phylogeny as a Pioneering Study of the SINE Method
2.2. Cichlid Phylogeny
2.3. The Origin of Cetaceans
2.4. The Monophyly or Paraphyly of Toothed Whales
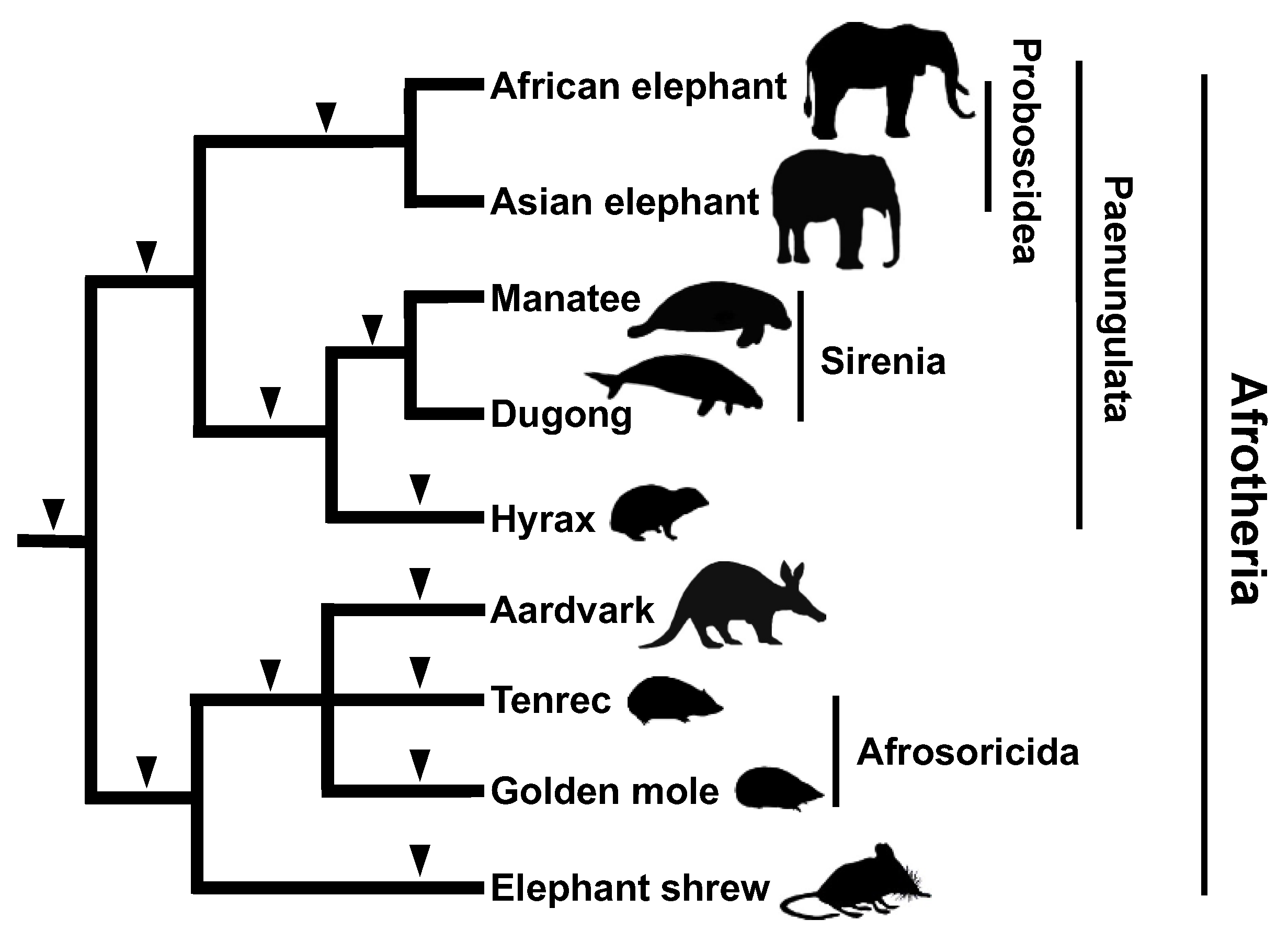
2.5. Afrotherian Phylogeny: Integration of Informatics Techniques
3. SINEs for Detecting Ancestral Polymorphisms and ILS
3.1. Inconsistent SINE Loci in Salmon, Cichlids, and Baleen Whales
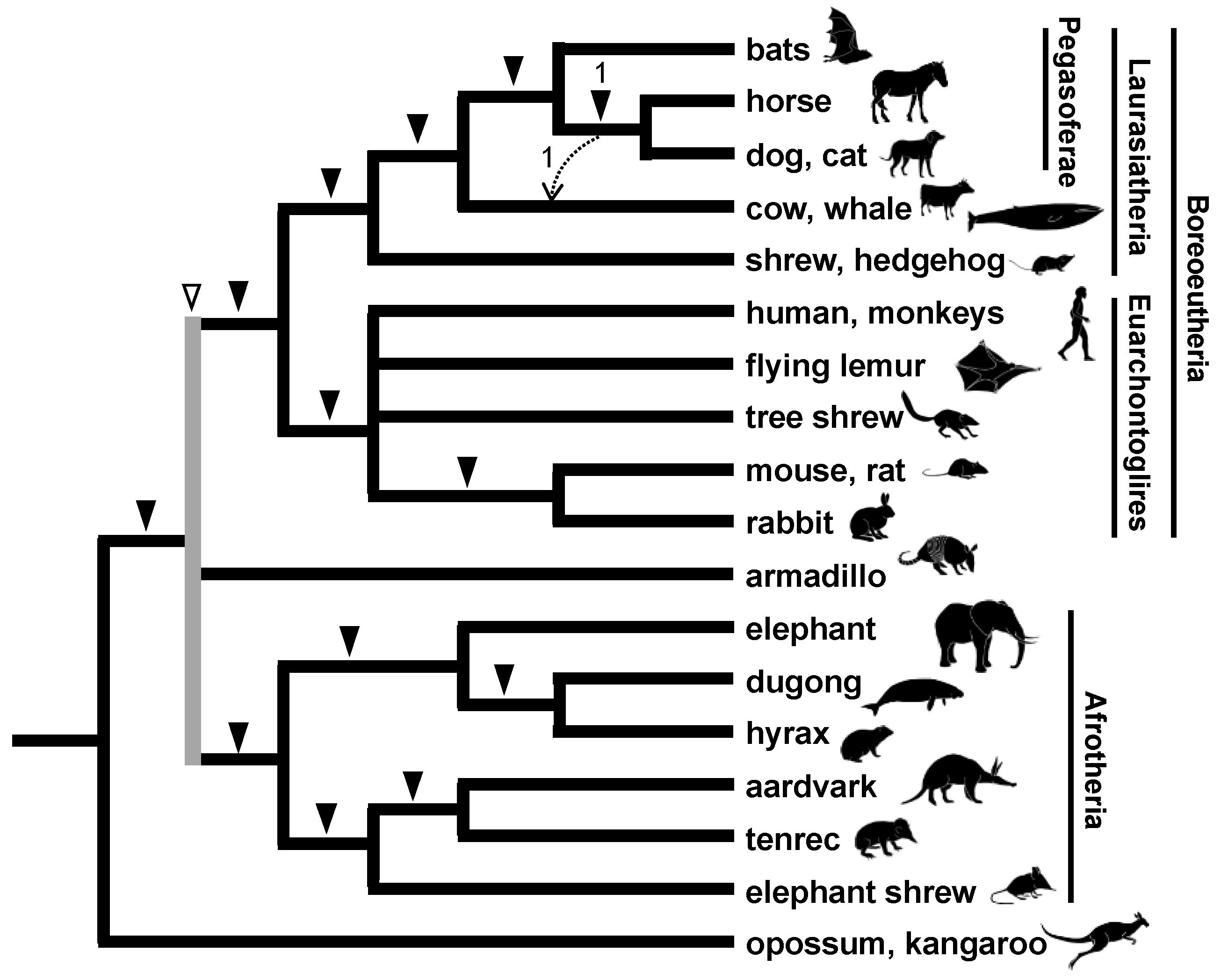
3.2. Interordinal Relationships among Eutherian Mammals
3.3. Rapid Divergence of the Three Major Eutherian Groups and an Association with the Continental Distribution
3.4. Advanced Retroposon Method with Next-Generation Sequencing Technology
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shedlock, A.M.; Okada, N. SINE insertions: Powerful tools for molecular systematics. Bioessays 2000, 22, 148–160. [Google Scholar] [CrossRef]
- Ohshima, K.; Hamada, M.; Terai, Y.; Okada, N. The 3’ ends of tRNA-derived short interspersed repetitive elements are derived from the 3’ ends of long interspersed repetitive elements. Mol. Cell. Biol. 1996, 16, 3756–3764. [Google Scholar] [CrossRef] [Green Version]
- Kajikawa, M.; Okada, N. LINEs mobilize SINEs in the eel through a shared 3’ sequence. Cell 2002, 111, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Kazazian, H.H.; Moran, J.V. The impact of L1 retrotransposons on the human genome. Nat. Genet. 1998, 19, 19–24. [Google Scholar] [CrossRef]
- Okada, N. SINEs: Short interspersed repeated elements of the eukaryotic genome. Trends Ecol. Evol. 1991, 6, 358–361. [Google Scholar] [CrossRef]
- Ullu, E.; Tschudi, C. Alu sequences are processed 7SL RNA genes. Nature 1984, 312, 171–172. [Google Scholar] [CrossRef]
- Rubin, C.M.; Houck, C.M.; Deininger, P.L.; Friedmann, T.; Schmid, C.W. Partial nucleotide sequence of the 300-nucleotide interspersed repeated human DNA sequences. Nature 1980, 284, 372–374. [Google Scholar] [CrossRef]
- Sakamoto, K.; Okada, N. Rodent type 2 Alu family, rat identifier sequence, rabbit C family, and bovine or goat 73-bp repeat may have evolved from tRNA genes. J. Mol. Evol. 1985, 22, 134–140. [Google Scholar] [CrossRef]
- Daniels, G.R.; Deininger, P.L. Repeat sequence families derived from mammalian tRNA genes. Nature 1985, 317, 819–822. [Google Scholar] [CrossRef]
- Ohshima, K.; Koishi, R.; Matsuo, M.; Okada, N. Several short interspersed repetitive elements (SINEs) in distant species may have originated from a common ancestral retrovirus: Characterization of a squid SINE and a possible mechanism for generation of tRNA-derived retroposons. Proc. Natl. Acad. Sci. USA 1993, 90, 6260–6264. [Google Scholar] [CrossRef] [Green Version]
- Endoh, H.; Okada, N. Total DNA transcription in vitro: A procedure to detect highly repetitive and transcribable sequences with tRNA-like structures. Proc. Natl. Acad. Sci. USA 1986, 83, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, K.; Okada, N. Generality of the tRNA origin of short interspersed repetitive elements (SINEs). Characterization of three different tRNA-derived retroposons in the octopus. J. Mol. Biol. 1994, 243, 25–37. [Google Scholar] [CrossRef]
- Matsumoto, K.; Murakami, K.; Okada, N. Gene for lysine tRNA1 may be a progenitor of the highly repetitive and transcribable sequences present in the salmon genome. Proc. Natl. Acad. Sci. USA 1986, 83, 3156–3160. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Terai, Y.; Nishida, M.; Okada, N. A novel family of short interspersed repetitive elements (SINEs) from cichlids: The patterns of insertion of SINEs at orthologous loci support the proposed monophyly of four major groups of cichlid fishes in Lake Tanganyika. Mol. Biol. Evol. 1998, 15, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, Y.; Matsumoto, S.; Kojima, S.; Ohshima, K.; Okada, N.; Machida, Y. Molecular characterization of a short interspersed repetitive element from tobacco that exhibits sequence homology to specific tRNAs. Proc. Natl. Acad. Sci. USA 1993, 90, 6562–6566. [Google Scholar] [CrossRef] [Green Version]
- Ray, D.A.; Xing, J.; Salem, A.H.; Batzer, M.A. SINEs of a nearly perfect character. Syst. Biol. 2006, 55, 928–935. [Google Scholar] [CrossRef]
- Nishihara, H.; Hasegawa, M.; Okada, N. Pegasoferae, an unexpected mammalian clade revealed by tracking ancient retroposon insertions. Proc. Natl. Acad. Sci. USA 2006, 103, 9929–9934. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, H.; Maruyama, S.; Okada, N. Retroposon analysis and recent geological data suggest near-simultaneous divergence of the three superorders of mammals. Proc. Natl. Acad. Sci. USA 2009, 106, 5235–5240. [Google Scholar] [CrossRef] [Green Version]
- Doronina, L.; Hughes, G.M.; Moreno-Santillan, D.; Lawless, C.; Lonergan, T.; Ryan, L.; Jebb, D.; Kirilenko, B.M.; Korstian, J.M.; Dávalos, L.M.; et al. Contradictory Phylogenetic Signals in the Laurasiatheria Anomaly Zone. Genes 2022, 13, 766. [Google Scholar] [CrossRef]
- Okada, N.; Shedlock, A.M.; Nikaido, M. Retroposon mapping in molecular systematics. Methods Mol. Biol. 2004, 260, 189–226. [Google Scholar]
- Murata, S.; Takasaki, N.; Saitoh, M.; Okada, N. Determination of the phylogenetic relationships among Pacific salmonids by using short interspersed elements (SINEs) as temporal landmarks of evolution. Proc. Natl. Acad. Sci. USA 1993, 90, 6995–6999. [Google Scholar] [CrossRef] [Green Version]
- Murata, S.; Takasaki, N.; Saitoh, M.; Tachida, H.; Okada, N. Details of retropositional genome dynamics that provide a rationale for a generic division: The distinct branching of all the pacific salmon and trout (Oncorhynchus) from the Atlantic salmon and trout (Salmo). Genetics 1996, 142, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, M.; Yasue, H.; Ohshima, K.; Abe, H.; Kato, H.; Kishiro, T.; Goto, M.; Munechika, I.; Okada, N. Molecular evidence from retroposons that whales form a clade within even-toed ungulates. Nature 1997, 388, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, M.; Rooney, A.P.; Okada, N. Phylogenetic relationships among cetartiodactyls based on insertions of short and long interpersed elements: Hippopotamuses are the closest extant relatives of whales. Proc. Natl. Acad. Sci. USA 1999, 96, 10261–10266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikaido, M.; Matsuno, F.; Hamilton, H.; Brownell, R.L.; Cao, Y.; Ding, W.; Zuoyan, Z.; Shedlock, A.M.; Fordyce, R.E.; Hasegawa, M.; et al. Retroposon analysis of major cetacean lineages: The monophyly of toothed whales and the paraphyly of river dolphins. Proc. Natl. Acad. Sci. USA 2001, 98, 7384–7389. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.; Ohme, M.; Zischler, H. SINE insertions in cladistic analyses and the phylogenetic affiliations of Tarsius bancanus to other primates. Genetics 2001, 157, 777–784. [Google Scholar] [CrossRef]
- Salem, A.H.; Ray, D.A.; Xing, J.; Callinan, P.A.; Myers, J.S.; Hedges, D.J.; Garber, R.K.; Witherspoon, D.J.; Jorde, L.B.; Batzer, M.A. Alu elements and hominid phylogenetics. Proc. Natl. Acad. Sci. USA 2003, 100, 12787–12791. [Google Scholar] [CrossRef] [Green Version]
- Roos, C.; Schmitz, J.; Zischler, H. Primate jumping genes elucidate strepsirrhine phylogeny. Proc. Natl. Acad. Sci. USA 2004, 101, 10650–10654. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, H.; Satta, Y.; Nikaido, M.; Thewissen, J.G.; Stanhope, M.J.; Okada, N. A retroposon analysis of Afrotherian phylogeny. Mol. Biol. Evol. 2005, 22, 1823–1833. [Google Scholar] [CrossRef]
- Kawai, K.; Nikaido, M.; Harada, M.; Matsumura, S.; Lin, L.K.; Wu, Y.; Hasegawa, M.; Okada, N. Intra- and interfamily relationships of Vespertilionidae inferred by various molecular markers including SINE insertion data. J. Mol. Evol. 2002, 55, 284–301. [Google Scholar] [CrossRef]
- Farwick, A.; Jordan, U.; Fuellen, G.; Huchon, D.; Catzeflis, F.; Brosius, J.; Schmitz, J. Automated scanning for phylogenetically informative transposed elements in rodents. Syst. Biol. 2006, 55, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Munemasa, M.; Nikaido, M.; Nishihara, H.; Donnellan, S.; Austin, C.C.; Okada, N. Newly discovered young CORE-SINEs in marsupial genomes. Gene 2008, 407, 176–185. [Google Scholar] [CrossRef]
- Nilsson, M.A.; Churakov, G.; Sommer, M.; Tran, N.V.; Zemann, A.; Brosius, J.; Schmitz, J. Tracking marsupial evolution using archaic genomic retroposon insertions. PLoS Biol. 2010, 8, e1000436. [Google Scholar] [CrossRef]
- Sasaki, T.; Takahashi, K.; Nikaido, M.; Miura, S.; Yasukawa, Y.; Okada, N. First application of the SINE (short interspersed repetitive element) method to infer phylogenetic relationships in reptiles: An example from the turtle superfamily Testudinoidea. Mol. Biol. Evol. 2004, 21, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, T.; Yasukawa, Y.; Takahashi, K.; Miura, S.; Shedlock, A.M.; Okada, N. Extensive morphological convergence and rapid radiation in the evolutionary history of the family Geoemydidae (old world pond turtles) revealed by SINE insertion analysis. Syst. Biol. 2006, 55, 912–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Nikaido, M.; Tsuda, T.T.; Inoko, H.; Mindell, D.P.; Murata, K.; Okada, N. The rise and fall of the CR1 subfamily in the lineage leading to penguins. Gene 2006, 365, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, T.; Nishihara, H.; Watanabe, M.; Okada, N. Determining the Position of Storks on the Phylogenetic Tree of Waterbirds by Retroposon Insertion Analysis. Genome Biol. Evol. 2015, 7, 3180–3189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shedlock, A.M.; Takahashi, K.; Okada, N. SINEs of speciation: Tracking lineages with retroposons. Trends Ecol. Evol. 2004, 19, 545–553. [Google Scholar] [CrossRef]
- Kuritzin, A.; Kischka, T.; Schmitz, J.; Churakov, G. Incomplete Lineage Sorting and Hybridization Statistics for Large-Scale Retroposon Insertion Data. PLoS Comput. Biol. 2016, 12, e1004812. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, M.; Hamilton, H.; Makino, H.; Sasaki, T.; Takahashi, K.; Goto, M.; Kanda, N.; Pastene, L.A.; Okada, N. Baleen whale phylogeny and a past extensive radiation event revealed by SINE insertion analysis. Mol. Biol. Evol. 2006, 23, 866–873. [Google Scholar] [CrossRef]
- Kido, Y.; Aono, M.; Yamaki, T.; Matsumoto, K.; Murata, S.; Saneyoshi, M.; Okada, N. Shaping and reshaping of salmonid genomes by amplification of tRNA-derived retroposons during evolution. Proc. Natl. Acad. Sci. USA 1991, 88, 2326–2330. [Google Scholar] [CrossRef] [Green Version]
- Britten, R.J.; Baron, W.F.; Stout, D.B.; Davidson, E.H. Sources and evolution of human Alu repeated sequences. Proc. Natl. Acad. Sci. USA 1988, 85, 4770–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasaki, N.; Murata, S.; Saitoh, M.; Kobayashi, T.; Park, L.; Okada, N. Species-specific amplification of tRNA-derived short interspersed repetitive elements (SINEs) by retroposition: A process of parasitization of entire genomes during the evolution of salmonids. Proc. Natl. Acad. Sci. USA 1994, 91, 10153–10157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, S.; Takasaki, N.; Okazaki, T.; Kobayashi, T.; Numachi, K.; Chang, K.-H.; Okada, N. Molecular evidence from short interspersed elements (SINEs) that Oncorhynchus masou (cherry salmon) is monophyletic. Can. J. Fish. Aquat. Sci. 1998, 55, 1864–1870. [Google Scholar] [CrossRef]
- Takasaki, N.; Park, L.; Kaeriyama, M.; Gharrett, A.J.; Okada, N. Characterization of species-specifically amplified SINEs in three salmonid species--chum salmon, pink salmon, and kokanee: The local environment of the genome may be important for the generation of a dominant source gene at a newly retroposed locus. J. Mol. Evol. 1996, 42, 103–116. [Google Scholar] [CrossRef]
- Takasaki, N.; Yamaki, T.; Hamada, M.; Park, L.; Okada, N. The salmon SmaI family of short interspersed repetitive elements (SINEs): Interspecific and intraspecific variation of the insertion of SINEs in the genomes of chum and pink salmon. Genetics 1997, 146, 369–380. [Google Scholar] [CrossRef]
- Hamada, M.; Takasaki, N.; Reist, J.D.; DeCicco, A.L.; Goto, A.; Okada, N. Detection of the ongoing sorting of ancestrally polymorphic SINEs toward fixation or loss in populations of two species of charr during speciation. Genetics 1998, 150, 301–311. [Google Scholar] [CrossRef]
- Fryer, G.; Iles, T.D. The Cichlid Fishes of the Great Lakes of Africa: Their Biology and Evolution; Oliver & Boyd: Edinburgh, UK, 1972. [Google Scholar]
- Meyer, A.; Kocher, T.D.; Basasibwaki, P.; Wilson, A.C. Monophyletic origin of Lake Victoria cichlid fishes suggested by mitochondrial DNA sequences. Nature 1990, 347, 550–553. [Google Scholar] [CrossRef] [Green Version]
- Nishida, M. Lake Tanganyika as an evolutionary reservoir of old lineages of East African cichlid fishes: Inferences from allozyme data. Experientia 1991, 47, 974–979. [Google Scholar] [CrossRef]
- Terai, Y.; Takahashi, K.; Okada, N. SINE cousins: The 3’-end tails of the two oldest and distantly related families of SINEs are descended from the 3’ ends of LINEs with the same genealogical origin. Mol. Biol. Evol. 1998, 15, 1460–1471. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Nishida, M.; Yuma, M.; Okada, N. Retroposition of the AFC family of SINEs (short interspersed repetitive elements) before and during the adaptive radiation of cichlid fishes in Lake Malawi and related inferences about phylogeny. J. Mol. Evol. 2001, 53, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Terai, Y.; Takezaki, N.; Mayer, W.E.; Tichy, H.; Takahata, N.; Klein, J.; Okada, N. Phylogenetic relationships among East African haplochromine fish as revealed by short interspersed elements (SINEs). J. Mol. Evol. 2004, 58, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Terai, Y.; Nishida, M.; Okada, N. Phylogenetic relationships and ancient incomplete lineage sorting among cichlid fishes in Lake Tanganyika as revealed by analysis of the insertion of retroposons. Mol. Biol. Evol. 2001, 18, 2057–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagl, S.; Tichy, H.; Mayer, W.E.; Takahata, N.; Klein, J. Persistence of neutral polymorphisms in Lake Victoria cichlid fish. Proc. Natl. Acad. Sci. USA 1998, 95, 14238–14243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terai, Y.; Takahashi, K.; Nishida, M.; Sato, T.; Okada, N. Using SINEs to probe ancient explosive speciation: “hidden” radiation of African cichlids? Mol. Biol. Evol. 2003, 20, 924–930. [Google Scholar] [CrossRef] [Green Version]
- Thewissen, J.G.; Hussain, S.T. Origin of underwater hearing in whales. Nature 1993, 361, 444–445. [Google Scholar] [CrossRef]
- Novacek, M.J. Mammalian phylogeny: Shaking the tree. Nature 1992, 356, 121–125. [Google Scholar] [CrossRef]
- Graur, D.; Higgins, D.G. Molecular evidence for the inclusion of cetaceans within the order Artiodactyla. Mol. Biol. Evol. 1994, 11, 357–364. [Google Scholar]
- Hasegawa, M.; Adachi, J. Phylogenetic position of cetaceans relative to artiodactyls: Reanalysis of mitochondrial and nuclear sequences. Mol. Biol. Evol. 1996, 13, 710–717. [Google Scholar] [CrossRef] [Green Version]
- Shimamura, M.; Abe, H.; Nikaido, M.; Ohshima, K.; Okada, N. Genealogy of families of SINEs in cetaceans and artiodactyls: The presence of a huge superfamily of tRNA (Glu)-derived families of SINEs. Mol. Biol. Evol. 1999, 16, 1046–1060. [Google Scholar] [CrossRef] [Green Version]
- Gatesy, J.; Hayashi, C.; Cronin, M.A.; Arctander, P. Evidence from milk casein genes that cetaceans are close relatives of hippopotamid artiodactyls. Mol. Biol. Evol. 1996, 13, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Gatesy, J. More DNA support for a Cetacea/Hippopotamidae clade: The blood-clotting protein gene gamma-fibrinogen. Mol. Biol. Evol. 1997, 14, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milinkovitch, M.; Thewissen, J. Even-toed fingerprints on whale ancestry. Nature 1997, 388, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Thewissen, J.G.; Williams, E.M.; Roe, L.J.; Hussain, S.T. Skeletons of terrestrial cetaceans and the relationship of whales to artiodactyls. Nature 2001, 413, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J.H.; Theodor, J.M. Hippopotamus and whale phylogeny. Nature 2009, 458, E1–E4. [Google Scholar] [CrossRef] [PubMed]
- Fordyce, R.E.; Barnes, L.G. The Evolutionary History of Whales and Dolphins. Annu. Rev. Earth Planet. Sci. 1994, 22, 419–455. [Google Scholar] [CrossRef]
- Rice, D.W. Marine Mammals of the World: Systematics and Distribution; Society for Marine Mammalogy: Lawrence, KS, USA, 1998. [Google Scholar]
- Milinkovitch, M.C.; Ortí, G.; Meyer, A. Revised phylogeny of whales suggested by mitochondrial ribosomal DNA sequences. Nature 1993, 361, 346–348. [Google Scholar] [CrossRef] [Green Version]
- Arnason, U.; Gullberg, A. Cytochrome b nucleotide sequences and the identification of five primary lineages of extant cetaceans. Mol. Biol. Evol. 1996, 13, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Adachi, J.; Hasegawa, M. Phylogeny of whales: Dependence of the inference on species sampling. Mol. Biol. Evol. 1995, 12, 177–179. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Adachi, J.; Milinkovitch, M.C. Novel phylogeny of whales supported by total molecular evidence. J. Mol. Evol. 1997, 44, S117–S120. [Google Scholar] [CrossRef]
- Nikaido, M.; Matsuno, F.; Abe, H.; Shimamura, M.; Hamilton, H.; Matsubayashi, H.; Okada, N. Evolution of CHR-2 SINEs in cetartiodactyl genomes: Possible evidence for the monophyletic origin of toothed whales. Mamm. Genome 2001, 12, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, M.; Piskurek, O.; Okada, N. Toothed whale monophyly reassessed by SINE insertion analysis: The absence of lineage sorting effects suggests a small population of a common ancestral species. Mol. Phylogenet. Evol. 2007, 43, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Nikaido, M.; Wada, S.; Yamada, T.K.; Cao, Y.; Hasegawa, M.; Okada, N. Balaenoptera omurai is a newly discovered baleen whale that represents an ancient evolutionary lineage. Mol. Phylogenet. Evol. 2006, 41, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.S.; Cleven, G.C.; Madsen, O.; de Jong, W.W.; Waddell, V.G.; Amrine, H.M.; Stanhope, M.J. Endemic African mammals shake the phylogenetic tree. Nature 1997, 388, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Novacek, M.J. Mammalian phylogeny: Genes and supertrees. Curr. Biol. 2001, 11, R573–R575. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, M.; Nishihara, H.; Fukumoto, Y.; Okada, N. Ancient SINEs from African endemic mammals. Mol. Biol. Evol. 2003, 20, 522–527. [Google Scholar] [CrossRef]
- Waddell, P.J.; Okada, N.; Hasegawa, M. Towards resolving the interordinal relationships of placental mammals. Syst. Biol. 1999, 48, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Murphy, W.J.; Eizirik, E.; O’Brien, S.J.; Madsen, O.; Scally, M.; Douady, C.J.; Teeling, E.; Ryder, O.A.; Stanhope, M.J.; de Jong, W.W.; et al. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science 2001, 294, 2348–2351. [Google Scholar] [CrossRef]
- Nishihara, H.; Okada, N.; Hasegawa, M. Rooting the eutherian tree: The power and pitfalls of phylogenomics. Genome Biol. 2007, 8, R199. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, H.; Okada, N. Retroposons: Genetic footprints on the evolutionary paths of life. Methods Mol. Biol. 2008, 422, 201–225. [Google Scholar]
- Kriegs, J.O.; Churakov, G.; Kiefmann, M.; Jordan, U.; Brosius, J.; Schmitz, J. Retroposed elements as archives for the evolutionary history of placental mammals. PLoS Biol. 2006, 4, e91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Cowled, C.; Shi, Z.; Huang, Z.; Bishop-Lilly, K.A.; Fang, X.; Wynne, J.W.; Xiong, Z.; Baker, M.L.; Zhao, W.; et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 2013, 339, 456–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doronina, L.; Churakov, G.; Kuritzin, A.; Shi, J.; Baertsch, R.; Clawson, H.; Schmitz, J. Speciation network in Laurasiatheria: Retrophylogenomic signals. Genome Res. 2017, 27, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Smith, D.G.; Funnell, B.M. Atlas of Cenozoic and Mesozoic Coastlines; Cambridge University Press: Cambridge, UK, 1994. [Google Scholar]
- Churakov, G.; Kriegs, J.O.; Baertsch, R.; Zemann, A.; Brosius, J.; Schmitz, J. Mosaic retroposon insertion patterns in placental mammals. Genome Res. 2009, 19, 868–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackett, S.J.; Kimball, R.T.; Reddy, S.; Bowie, R.C.; Braun, E.L.; Braun, M.J.; Chojnowski, J.L.; Cox, W.A.; Han, K.-L.; Harshman, J.; et al. A phylogenomic study of birds reveals their evolutionary history. Science 2008, 320, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Kimball, R.T.; Wang, N.; Heimer-McGinn, V.; Ferguson, C.; Braun, E.L. Identifying localized biases in large datasets: A case study using the avian tree of life. Mol. Phylogenet. Evol. 2013, 69, 1021–1032. [Google Scholar] [CrossRef]
- Springer, M.S.; Molloy, E.K.; Sloan, D.B.; Simmons, M.P.; Gatesy, J. ILS-Aware Analysis of Low-Homoplasy Retroelement Insertions: Inference of Species Trees and Introgression Using Quartets. J. Hered. 2020, 111, 147–168. [Google Scholar] [CrossRef] [Green Version]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Stiller, J.; Deng, Y.; Armstrong, J.; Fang, Q.; Reeve, A.H.; Xie, D.; Chen, G.; Guo, C.; Faircloth, B.C.; et al. Dense sampling of bird diversity increases power of comparative genomics. Nature 2020, 587, 252–257. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikaido, M.; Nishihara, H.; Okada, N. SINEs as Credible Signs to Prove Common Ancestry in the Tree of Life: A Brief Review of Pioneering Case Studies in Retroposon Systematics. Genes 2022, 13, 989. https://doi.org/10.3390/genes13060989
Nikaido M, Nishihara H, Okada N. SINEs as Credible Signs to Prove Common Ancestry in the Tree of Life: A Brief Review of Pioneering Case Studies in Retroposon Systematics. Genes. 2022; 13(6):989. https://doi.org/10.3390/genes13060989
Chicago/Turabian StyleNikaido, Masato, Hidenori Nishihara, and Norihiro Okada. 2022. "SINEs as Credible Signs to Prove Common Ancestry in the Tree of Life: A Brief Review of Pioneering Case Studies in Retroposon Systematics" Genes 13, no. 6: 989. https://doi.org/10.3390/genes13060989
APA StyleNikaido, M., Nishihara, H., & Okada, N. (2022). SINEs as Credible Signs to Prove Common Ancestry in the Tree of Life: A Brief Review of Pioneering Case Studies in Retroposon Systematics. Genes, 13(6), 989. https://doi.org/10.3390/genes13060989