
Characterization of Expression and Epigenetic Features of Core Genes in Common Wheat



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of the Core Genome of Common Wheat
2.2. Gene Expression Patterns and Epigenetic Landscapes of Core Genes
2.3. Co-Expression Analyses and Construction of Gene Regulatory Networks
2.4. Statistical Analyses
3. Results
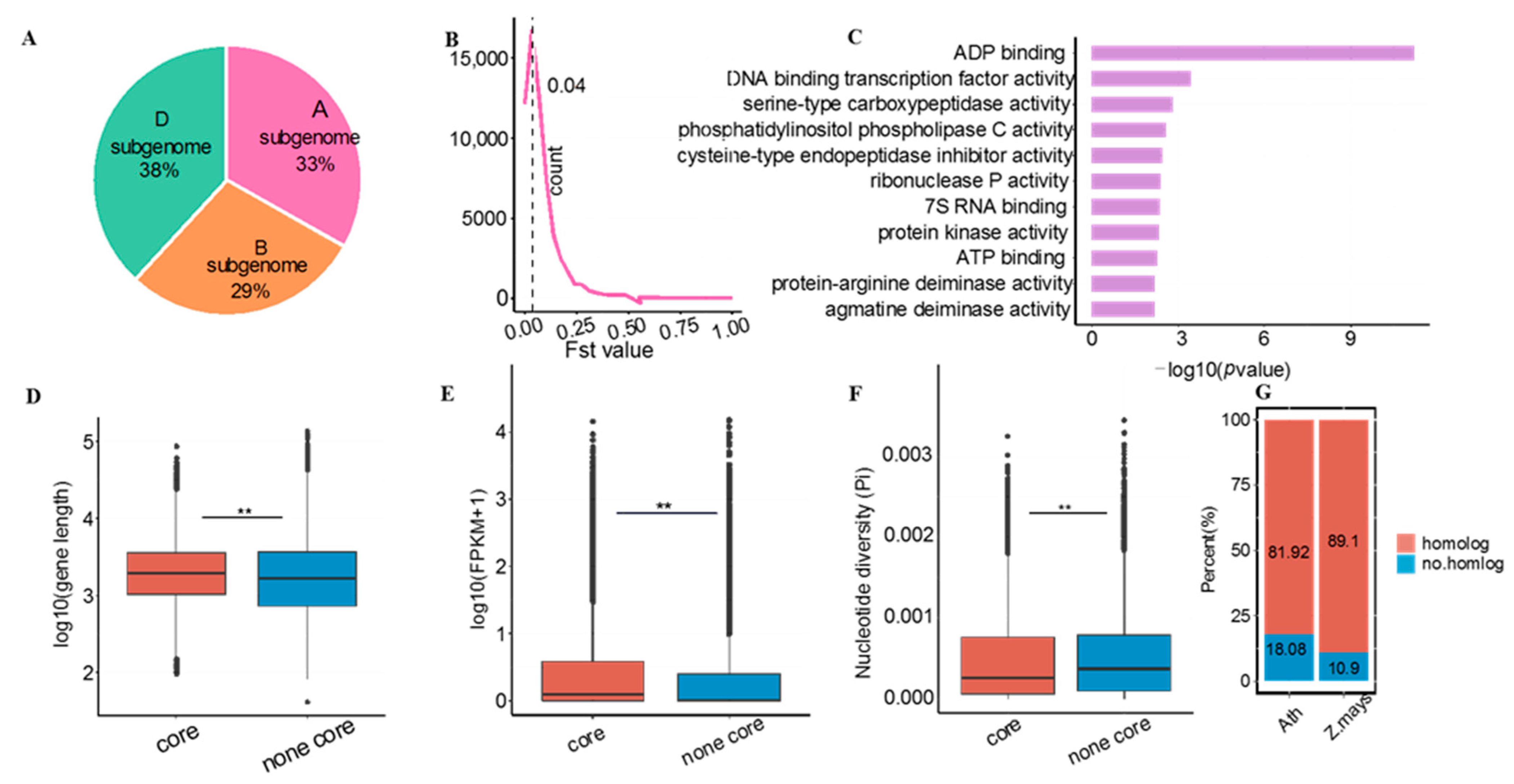
3.1. Identification of the Core Genome of Common Wheat
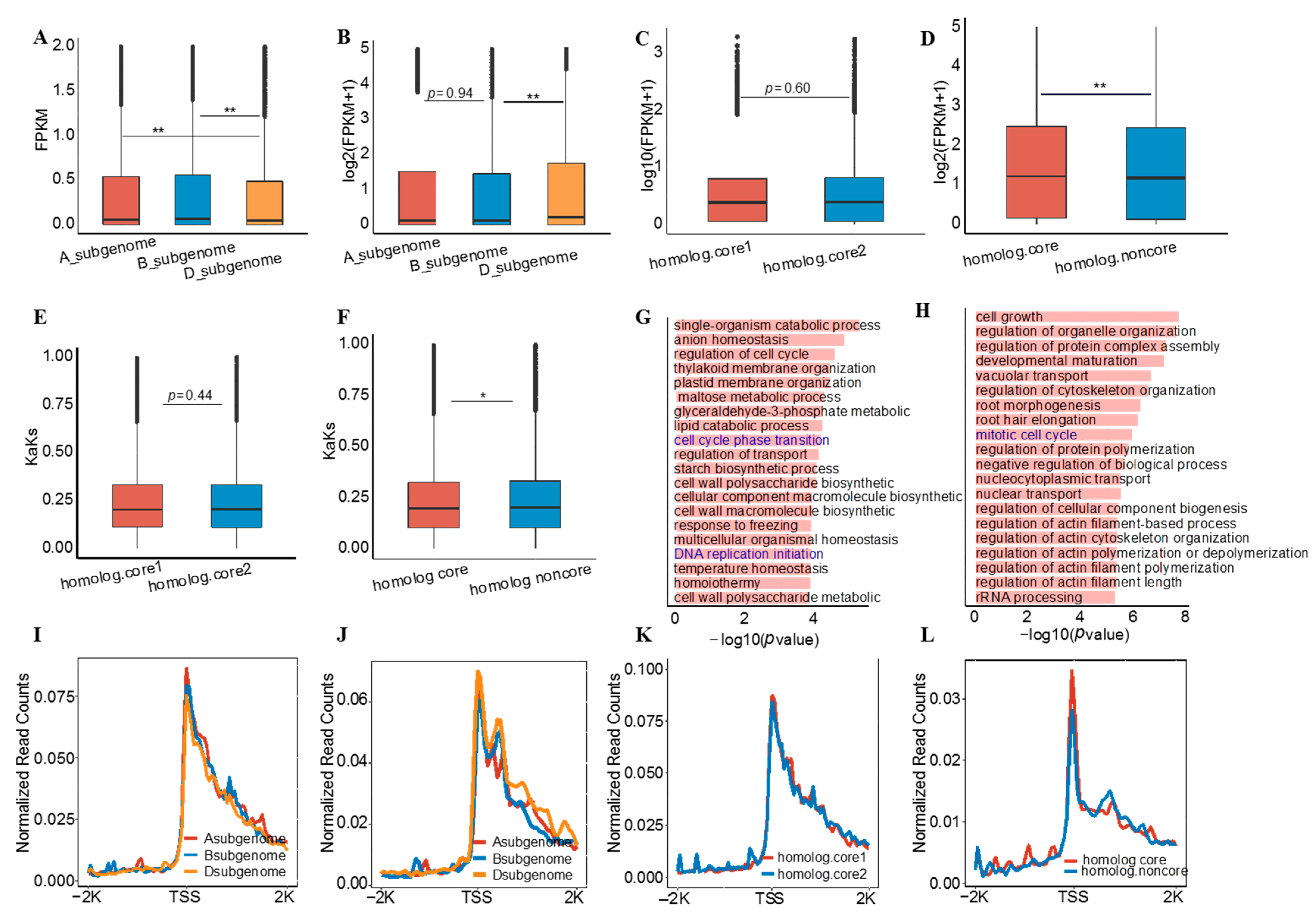
3.2. Core and Non-Core Genes Have Different Roles in Subgenome Differentiation
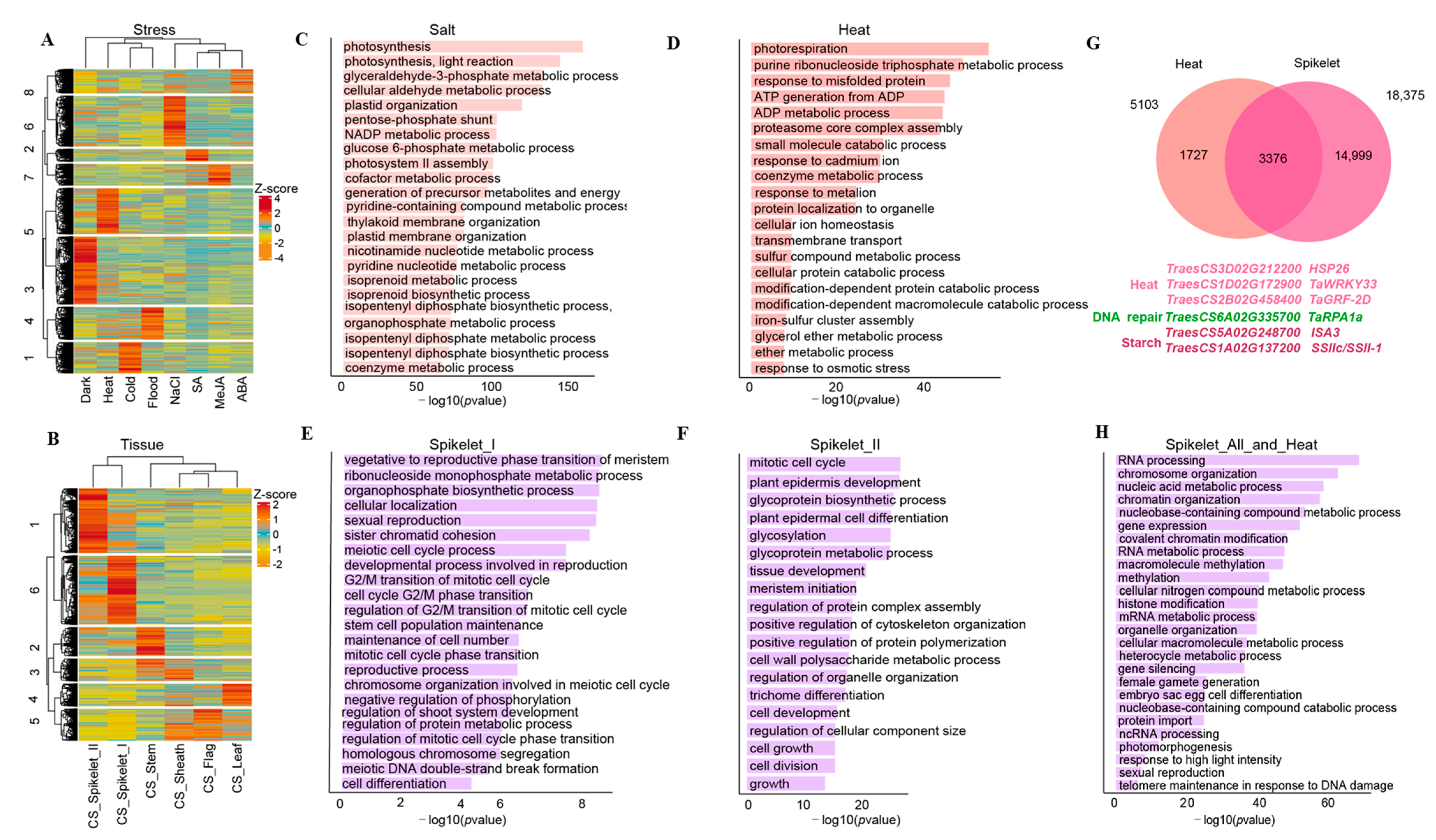
3.3. Potential Biological Relevance of Core Genes in Wheat
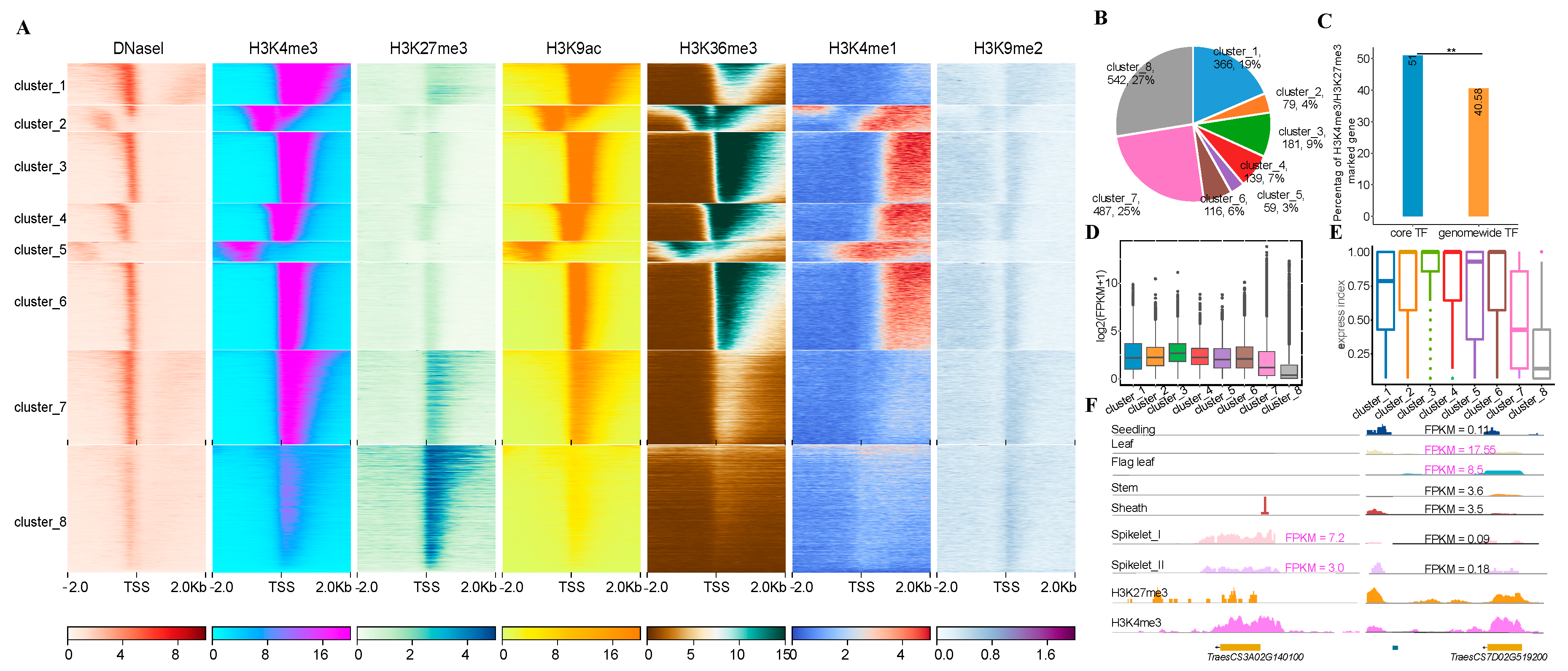
3.4. Core Transcription Factors Enriched with H3K27me3 and H3K4me3
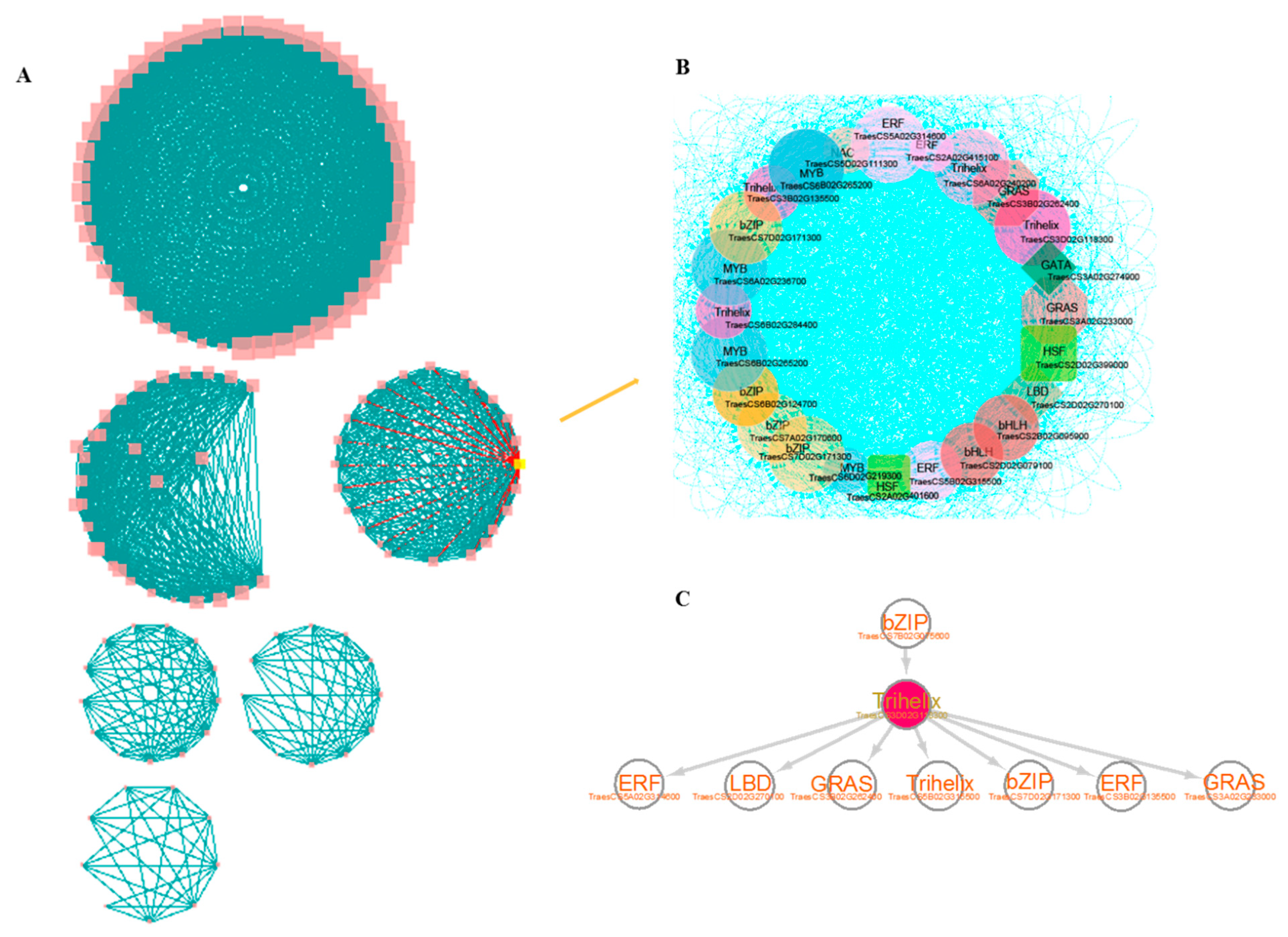
3.5. Construction of Core TF-Centered Regulatory Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ballouz, S.; Dobin, A.; Gillis, J.A. Is it time to change the reference genome? Genome Biol. 2019, 20, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au, K.F.; Valiente-Mullor, C.; Beamud, B.; Ansari, I.; Francés-Cuesta, C.; García-González, N.; Mejía, L.; Ruiz-Hueso, P.; González-Candelas, F. One is not enough: On the effects of reference genome for the mapping and subsequent analyses of short-reads. PLoS Comput. Biol. 2021, 17, e1008678. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, R.M.; Salzberg, S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558.e3516. [Google Scholar] [CrossRef]
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.S.; Gundlach, H.; Monat, C.; Lux, T.; Kamal, N.; Lang, D.; Himmelbach, A.; et al. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289. [Google Scholar] [CrossRef]
- Hufford, M.B.; Seetharam, A.S.; Woodhouse, M.R.; Chougule, K.M.; Ou, S.; Liu, J.; Ricci, W.A.; Guo, T.; Olson, A.; Qiu, Y.; et al. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 2021, 373, 655–662. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.-A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176.e113. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.P.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.P.; Contreras-Moreira, B.; Woods, D.P.; Des Marais, D.L.; Burgess, D.; Shu, S.; Stritt, C.; Roulin, A.C.; Schackwitz, W.; Tyler, L.; et al. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat. Commun. 2017, 8, 2184. [Google Scholar] [CrossRef]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.K.K.; Tirnaz, S.; Dolatabadian, A.; Schiessl, S.V.; Samans, B.; Montenegro, J.D.; Parkin, I.A.P.; et al. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2018, 16, 1265–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yuan, D.; Wang, P.; Wang, Q.; Sun, M.; Liu, Z.; Si, H.; Xu, Z.; Ma, Y.; Zhang, B.; et al. Cotton pan-genome retrieves the lost sequences and genes during domestication and selection. Genome Biol. 2021, 22, 119. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Edger, P.P.; Xue, L.; Qiong, L.; Lu, J.; Zhang, Y.; Cao, Q.; Yocca, A.E.; Platts, A.E.; Knapp, S.J.; et al. Evolutionary history and pan-genome dynamics of strawberry (Fragaria spp.). Proc. Natl. Acad. Sci. USA 2021, 118, e2105431118. [Google Scholar] [CrossRef]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Della Coletta, R.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 3. [Google Scholar] [CrossRef]
- Tao, Y.; Zhao, X.; Mace, E.; Henry, R.; Jordan, D. Exploring and Exploiting Pan-genomics for Crop Improvement. Mol. Plant 2019, 12, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Marroni, F.; Pinosio, S.; Morgante, M. Structural variation and genome complexity: Is dispensable really dispensable? Curr. Opin. Plant Biol. 2014, 18, 31–36. [Google Scholar] [CrossRef]
- Maddamsetti, R.; Hatcher, P.J.; Green, A.G.; Williams, B.L.; Marks, D.S.; Lenski, R.E. Core Genes Evolve Rapidly in the Long-Term Evolution Experiment with Escherichia coli. Genome Biol. Evol. 2017, 9, 1072–1083. [Google Scholar] [CrossRef]
- Li, Y.-H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.-G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Shiferaw, B.; Smale, M.; Braun, H.-J.; Duveiller, E.; Reynolds, M.; Muricho, G. Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Secur. 2013, 5, 291–317. [Google Scholar] [CrossRef] [Green Version]
- Wicker, T.; Gundlach, H.; Spannagl, M.; Uauy, C.; Borrill, P.; Ramirez-Gonzalez, R.H.; De Oliveira, R.; International Wheat Genome Sequencing, C.; Mayer, K.F.X.; Paux, E.; et al. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 2018, 19, 103. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, J.D.; Golicz, A.A.; Bayer, P.E.; Hurgobin, B.; Lee, H.; Chan, C.K.K.; Visendi, P.; Lai, K.; Dolezel, J.; Batley, J.; et al. The pangenome of hexaploid bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, C.Y.; Jiao, C.Z.; Hou, J.; Li, T.; Liu, H.X.; Wang, Y.Q.; Zheng, J.; Liu, H.; Bi, Z.H.; Xu, F.F.; et al. Resequencing of 145 Landmark Cultivars Reveals Asymmetric Sub-genome Selection and Strong Founder Genotype Effects on Wheat Breeding in China. Mol. Plant 2020, 13, 1733–1751. [Google Scholar] [CrossRef]
- Sato, K.; Abe, F.; Mascher, M.; Haberer, G.; Gundlach, H.; Spannagl, M.; Shirasawa, K.; Isobe, S. Chromosome-scale genome assembly of the transformation-amenable common wheat cultivar ‘Fielder’. DNA Res. 2021, 28, dsab008. [Google Scholar] [CrossRef]
- Walkowiak, S.; Gao, L.; Monat, C.; Haberer, G.; Kassa, M.T.; Brinton, J.; Ramirez-Gonzalez, R.H.; Kolodziej, M.C.; Delorean, E.; Thambugala, D.; et al. Multiple wheat genomes reveal global variation in modern breeding. Nature 2020, 588, 277–283. [Google Scholar] [CrossRef]
- Brinton, J.; Ramirez-Gonzalez, R.H.; Simmonds, J.; Wingen, L.; Orford, S.; Griffiths, S.; Haberer, G.; Spannagl, M.; Walkowiak, S.; Pozniak, C.; et al. A haplotype-led approach to increase the precision of wheat breeding. Commun. Biol. 2020, 3, 712. [Google Scholar] [CrossRef]
- Harris, C.D.; Torrance, E.L.; Raymann, K.; Bobay, L.-M.; Ouangraoua, A. CoreCruncher: Fast and Robust Construction of Core Genomes in Large Prokaryotic Data Sets. Mol. Biol. Evol. 2021, 38, 727–734. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.Y.; Li, Z.J.; Zhang, Y.E.; Zhang, Y.Y.; Xie, Y.L.; Ye, L.H.; Zhuang, Y.L.; Lin, K.D.; Zhao, F.; Guo, J.Y.; et al. An atlas of wheat epigenetic regulatory elements reveals subgenome divergence in the regulation of development and stress responses. Plant Cell 2021, 33, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fu, W.; Wang, R.; Hu, D.; Cheng, H.; Zhao, J.; Jiang, Y.; Kang, Z. WGVD: An integrated web-database for wheat genome variation and selective signatures. Database 2020, 2020, baaa090. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [Green Version]
- Isalan, M.; Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Zhu, Q.; Gao, S.; Zhang, W. Identification of Key Transcription Factors Related to Bacterial Spot Resistance in Pepper through Regulatory Network Analyses. Genes 2021, 12, 1351. [Google Scholar] [CrossRef]
- IWGSC. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 661. [Google Scholar] [CrossRef] [Green Version]
- Kronforst, M.R.; Fleming, T.H. Lack of genetic differentiation among widely spaced subpopulations of a butterfly with home range behaviour. Heredity 2001, 86, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.Z.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Blanc-Mathieu, R.; GrandVuillemin, L.; Huang, Y.; Stigliani, A.; Lucas, J.; Thévenon, E.; Loue-Manifel, J.; Turchi, L.; Daher, H.; et al. The LEAFY floral regulator displays pioneer transcription factor properties. Mol. Plant 2021, 14, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y. Evolution of Polyploid Triticum Wheats under Cultivation: The Role of Domestication, Natural Hybridization and Allopolyploid Speciation in their Diversification. Plant Cell Physiol. 2011, 52, 750–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Chen, Y.; Li, Z.; Zhu, J.; Liu, M.; Zhang, Y.; Dong, Z. Enhancer transcription detected in the nascent transcriptomic landscape of bread wheat. Genome Biol. 2022, 23, 109. [Google Scholar] [CrossRef]
- Seleiman, M.F.; Aslam, M.T.; Alhammad, B.A.; Hassan, M.U.; Maqbool, R.; Chattha, M.U.; Khan, I.; Gitari, H.I.; Uslu, O.S.; Roy, R.; et al. Salinity Stress in Wheat: Effects, Mechanisms and Management Strategies. Phyton-Int. J. Exp. Bot. 2022, 91, 667–694. [Google Scholar] [CrossRef]
- Akter, N.; Rafiqul Islam, M. Heat stress effects and management in wheat. A review. Agron. Sustain. Dev. 2017, 37, 37. [Google Scholar] [CrossRef]
- Saddiq, M.S.; Iqbal, S.; Hafeez, M.B.; Ibrahim, A.M.H.; Raza, A.; Fatima, E.M.; Baloch, H.; Jahanzaib; Woodrow, P.; Ciarmiello, L.F. Effect of Salinity Stress on Physiological Changes in Winter and Spring Wheat. Agronomy 2021, 11, 1193. [Google Scholar] [CrossRef]
- Djanaguiraman, M.; Narayanan, S.; Erdayani, E.; Prasad, P.V.V. Effects of high temperature stress during anthesis and grain filling periods on photosynthesis, lipids and grain yield in wheat. BMC Plant Biol. 2020, 20, 268. [Google Scholar] [CrossRef]
- Gao, X.Q.; Wang, N.; Wang, X.L.; Zhang, X.S. Architecture of Wheat Inflorescence: Insights from Rice. Trends Plant Sci. 2019, 24, 802–809. [Google Scholar] [CrossRef]
- Javelle, M.; Vernoud, V.; Rogowsky, P.M.; Ingram, G.C. Epidermis: The formation and functions of a fundamental plant tissue. New Phytol. 2010, 189, 17–39. [Google Scholar] [CrossRef]
- Harrison, C.J.; Alvey, E.; Henderson, I.R. Meiosis in flowering plants and other green organisms. J. Exp. Bot. 2010, 61, 2863–2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobell, D.B.; Hammer, G.L.; McLean, G.; Messina, C.; Roberts, M.J.; Schlenker, W. The critical role of extreme heat for maize production in the United States. Nat. Clim. Chang. 2013, 3, 497–501. [Google Scholar] [CrossRef]
- Farooq, M.; Bramley, H.; Palta, J.A.; Siddique, K.H.M. Heat Stress in Wheat during Reproductive and Grain-Filling Phases. Crit. Rev. Plant Sci. 2011, 30, 491–507. [Google Scholar] [CrossRef]
- Jagadish, S.V.K.; Craufurd, P.Q.; Wheeler, T.R. High temperature stress and spikelet fertility in rice (Oryza sativa L.). J. Exp. Bot. 2007, 58, 1627–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.-H.; Xu, J.-Y.; Wang, Y.-X.; Liu, J.-M.; Li, P.-S.; Chen, M.; Ma, Y.-Z.; Xu, Z.-S. Drought-responsive WRKY transcription factor genes TaWRKY1 and TaWRKY33 from wheat confer drought and/or heat resistance in Arabidopsis. BMC Plant Biol. 2016, 16, 116. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Khurana, N.; Chauhan, H.; Khurana, P. Wheat Chloroplast Targeted sHSP26 Promoter Confers Heat and Abiotic Stress Inducible Expression in Transgenic Arabidopsis Plants. PLoS ONE 2013, 8, 1912–1931. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Li, Z.; Wang, M.; Lin, K.; Xie, Y.; Guo, J.; Ye, L.; Zhuang, Y.; Teng, W.; Ran, X.; Tong, Y.; et al. The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements. Genome Biol. 2019, 20, 139. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Zhang, W.; Marand, A.P.; Zhu, B.; Buell, C.R.; Jiang, J. Cold stress induces enhanced chromatin accessibility and bivalent histone modifications H3K4me3 and H3K27me3 of active genes in potato. Genome Biol. 2019, 20, 123. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; González-Ramírez, M.; Alcaine-Colet, A.; Aranda, S.; Di Croce, L. The Bivalent Genome: Characterization, Structure, and Regulation. Trends Genet. 2020, 36, 118–131. [Google Scholar] [CrossRef]
- Kazemian, M.; Pham, H.; Wolfe, S.A.; Brodsky, M.H.; Sinha, S. Widespread evidence of cooperative DNA binding by transcription factors in Drosophila development. Nucleic Acids Res. 2013, 41, 8237–8252. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Ahmad, K.; Ramachandran, S. Cooperative binding between distant transcription factors is a hallmark of active enhancers. Mol. Cell 2021, 81, 1651–1665.e1654. [Google Scholar] [CrossRef] [PubMed]
- Wei, H. Construction of a hierarchical gene regulatory network centered around a transcription factor. Brief. Bioinform. 2019, 20, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-González, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361, eaar6089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukao, T.; Bailey-Serres, J. Submergence tolerance conferred by Sub1A is mediated by SLR1 and SLRL1 restriction of gibberellin responses in rice. Proc. Natl. Acad. Sci. USA 2008, 105, 16814–16819. [Google Scholar] [CrossRef] [Green Version]
- Itoh, H.; Shimada, A.; Ueguchi-Tanaka, M.; Kamiya, N.; Hasegawa, Y.; Ashikari, M.; Matsuoka, M. Overexpression of a GRAS protein lacking the DELLA domain confers altered gibberellin responses in rice. Plant J. 2005, 44, 669–679. [Google Scholar] [CrossRef]
- Qi, W.; Sun, F.; Wang, Q.; Chen, M.; Huang, Y.; Feng, Y.-Q.; Luo, X.; Yang, J. Rice Ethylene-Response AP2/ERF Factor OsEATB Restricts Internode Elongation by Down-Regulating a Gibberellin Biosynthetic Gene. Plant Physiol. 2011, 157, 216–228. [Google Scholar] [CrossRef] [Green Version]
- Bhanbhro, N.; Xiao, B.; Han, L.; Lu, H.; Wang, H.; Yang, C. Adaptive strategy of allohexaploid wheat to long-term salinity stress. BMC Plant Biol. 2020, 20, 210. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Zheng, H.; Luo, R.; Zhu, H.; Li, Q.; Qian, W.; Ren, Y.; Tian, G.; Li, J.; et al. Building the sequence map of the human pan-genome. Nat. Biotechnol. 2010, 28, 57–63. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Hübner, S.; Bercovich, N.; Todesco, M.; Mandel, J.R.; Odenheimer, J.; Ziegler, E.; Lee, J.S.; Baute, G.J.; Owens, G.L.; Grassa, C.J.; et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 2018, 5, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, L.; Zhang, X.; Yang, N.; Guo, J.; Wang, M.; Ji, S.; Zhao, X.; Yin, P.; Cai, L.; et al. Convergent selection of a WD40 protein that enhances grain yield in maize and rice. Science 2022, 375, eabg7985. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone Methylation in Higher Plants. Annu. Rev. Plant Biol. 2010, 61, 395–420. [Google Scholar] [CrossRef]
- Bastow, R.; Mylne, J.S.; Lister, C.; Lippman, Z.; Martienssen, R.A.; Dean, C. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 2004, 427, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Ramos-Cruz, D.; Becker, C. The role of plant epigenetics in biotic interactions. New Phytol. 2019, 221, 731–737. [Google Scholar] [CrossRef]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef]
- Chang, Y.N.; Zhu, C.; Jiang, J.; Zhang, H.; Zhu, J.K.; Duan, C.G. Epigenetic regulation in plant abiotic stress responses. J. Integr. Plant Biol. 2020, 62, 563–580. [Google Scholar] [CrossRef]
- Liu, N.; Fromm, M.; Avramova, Z. H3K27me3 and H3K4me3 Chromatin Environment at Super-Induced Dehydration Stress Memory Genes of Arabidopsis thaliana. Mol. Plant 2014, 7, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Berr, A.; McCallum, E.J.; Ménard, R.; Meyer, D.; Fuchs, J.; Dong, A.; Shen, W.-H. Arabidopsis SET DOMAIN GROUP2Is Required for H3K4 Trimethylation and Is Crucial for Both Sporophyte and Gametophyte Development. Plant Cell 2010, 22, 3232–3248. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, D.; Zhang, W. Characterization of Expression and Epigenetic Features of Core Genes in Common Wheat. Genes 2022, 13, 1112. https://doi.org/10.3390/genes13071112
Zheng D, Zhang W. Characterization of Expression and Epigenetic Features of Core Genes in Common Wheat. Genes. 2022; 13(7):1112. https://doi.org/10.3390/genes13071112
Chicago/Turabian StyleZheng, Dongyang, and Wenli Zhang. 2022. "Characterization of Expression and Epigenetic Features of Core Genes in Common Wheat" Genes 13, no. 7: 1112. https://doi.org/10.3390/genes13071112
APA StyleZheng, D., & Zhang, W. (2022). Characterization of Expression and Epigenetic Features of Core Genes in Common Wheat. Genes, 13(7), 1112. https://doi.org/10.3390/genes13071112