
Comparative Transcriptome Profiling Reveals the Genes Involved in Storage Root Expansion in Sweetpotato (Ipomoea batatas (L.) Lam.)

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. RNA Extraction, cDNA Library Construction and Transcriptome Analysis
2.3. De Novo Assembly and Data Processing
2.4. Differential Expression Analysis (DEGs)
2.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
2.6. Quantitative Real-Time PCR (qRT-PCR) Validation
3. Results
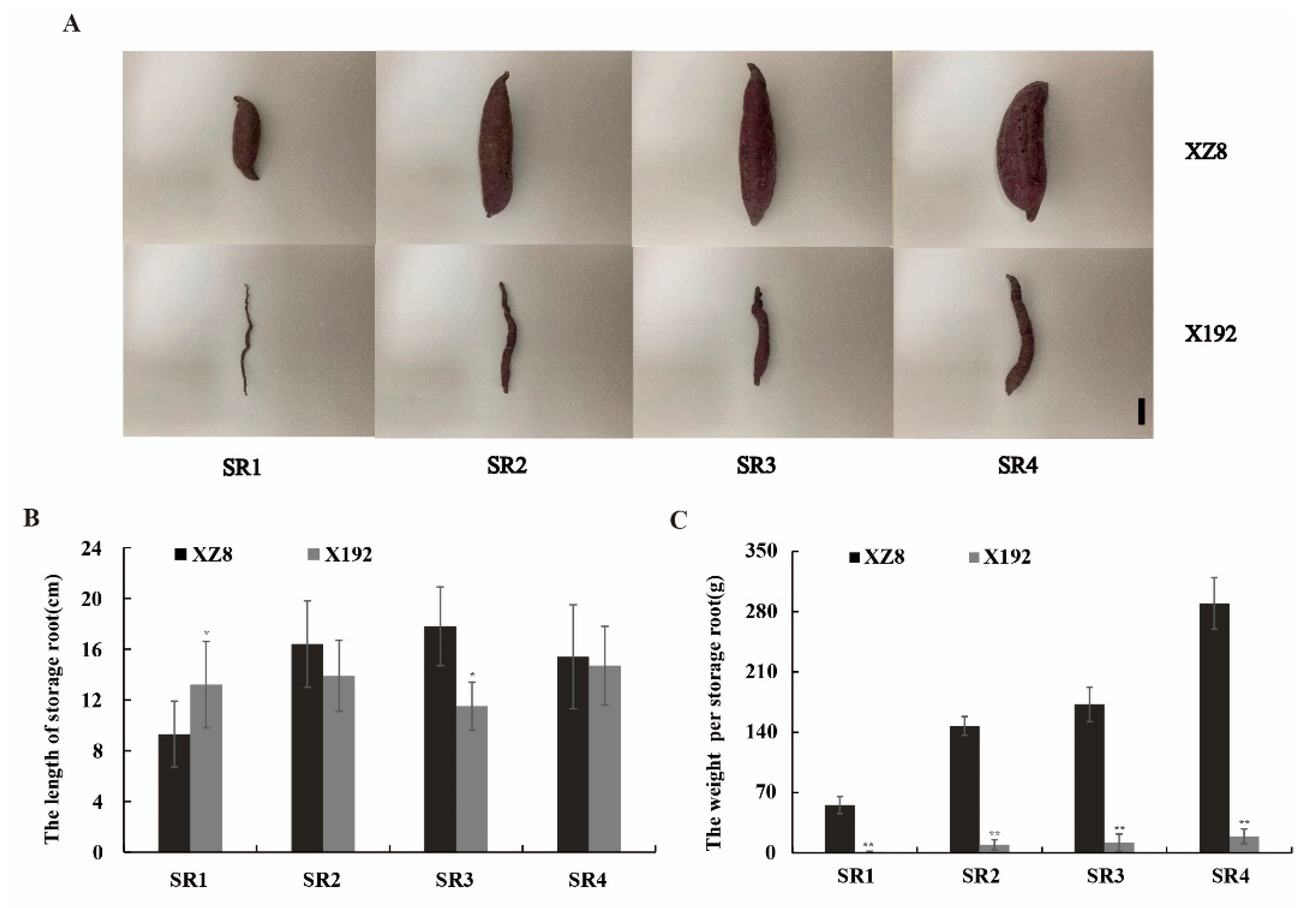
3.1. Sweetpotato SR Characteristics at Different Stages
3.2. RNA-Seq Analysis
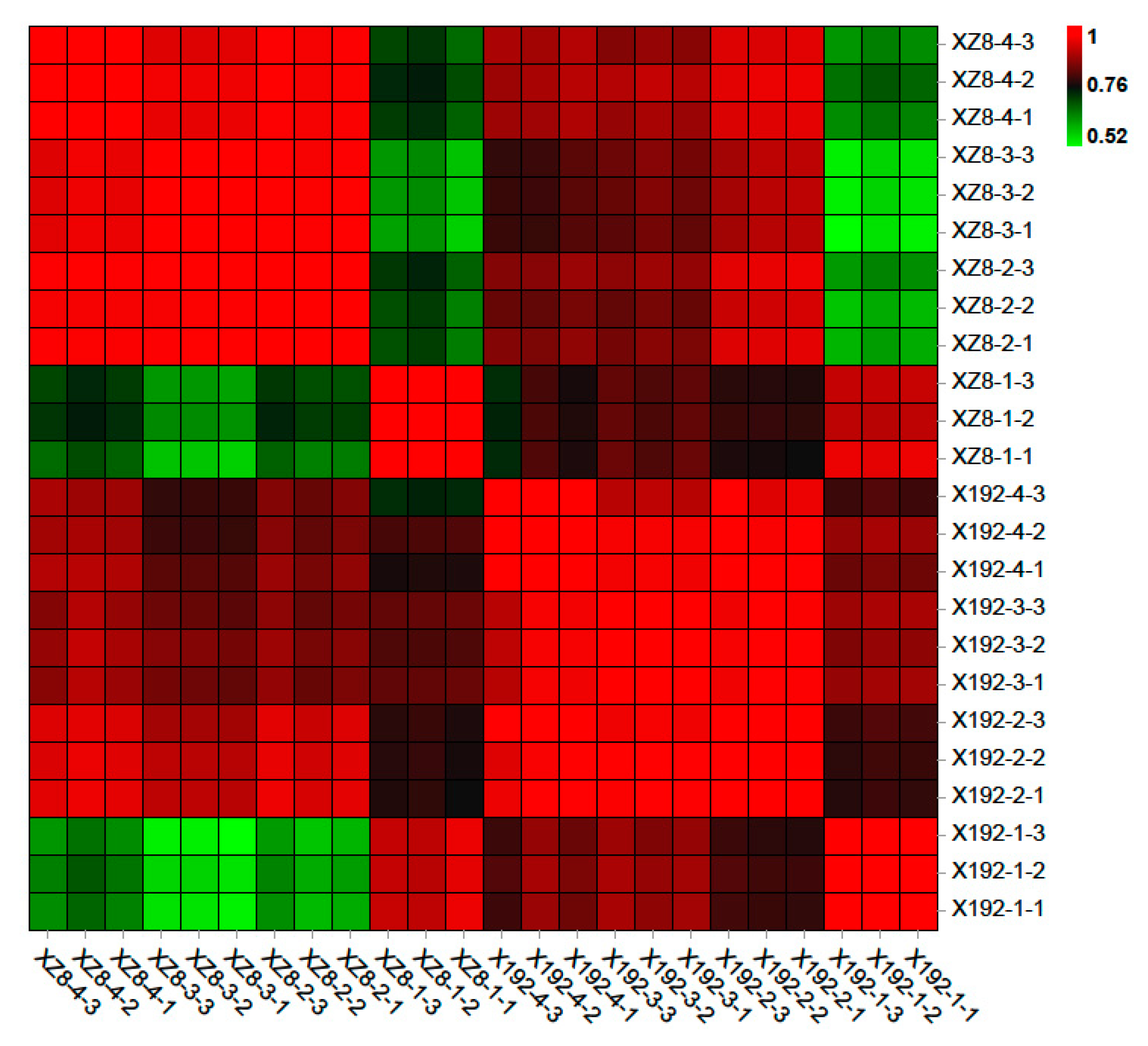
3.3. Identification of DEGs and Cluster Analysis
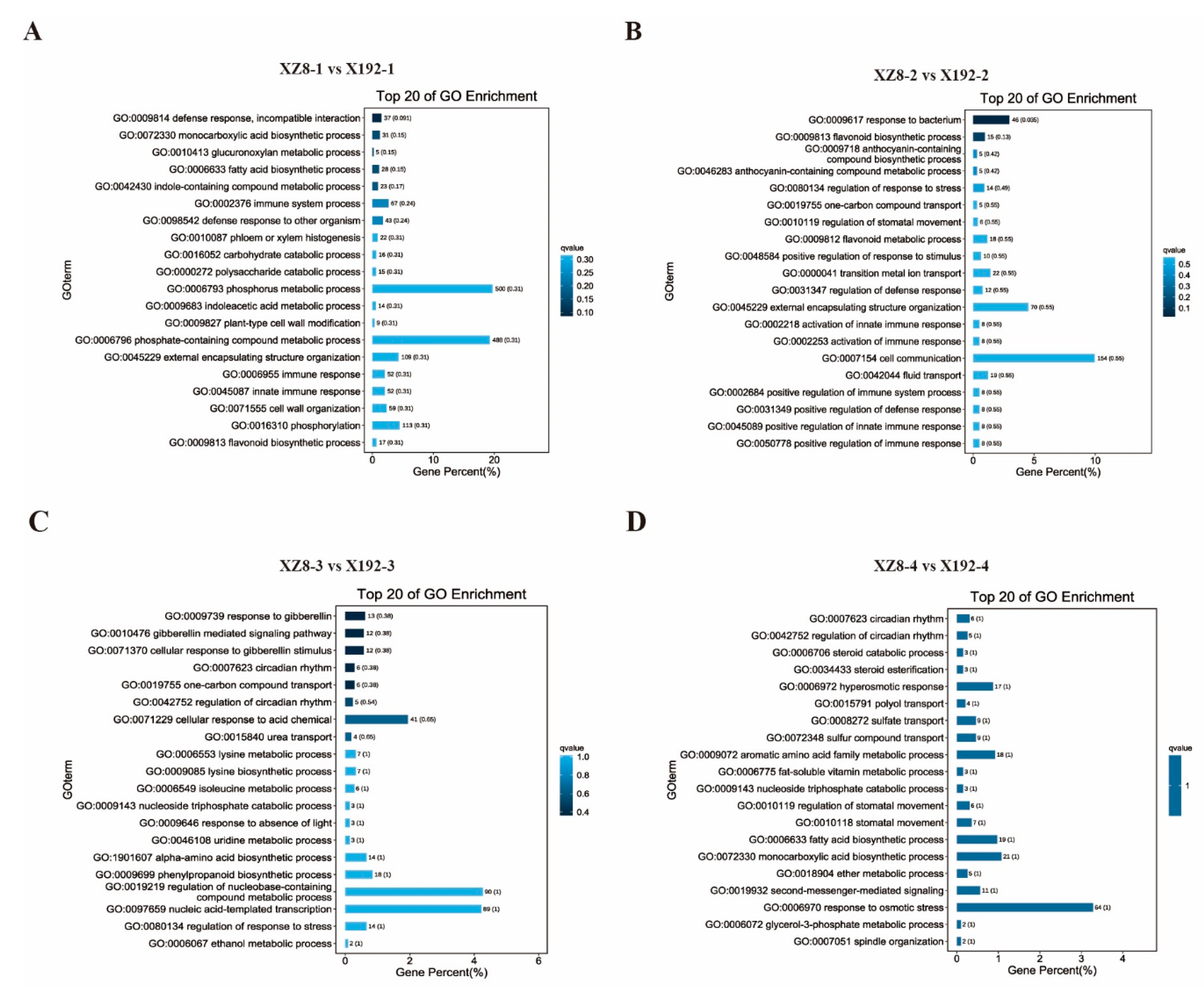
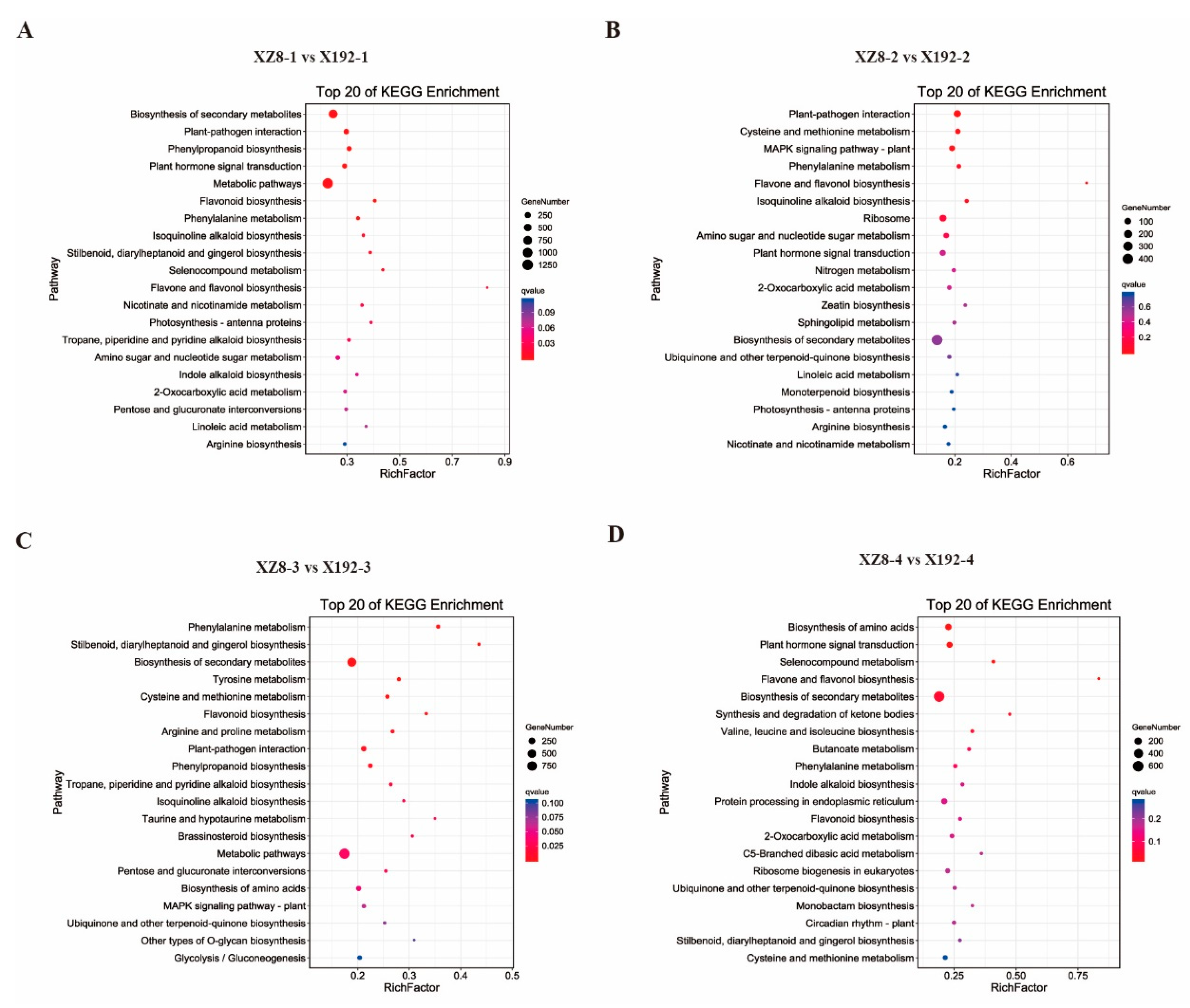
3.4. Gene Enrichment Analysis
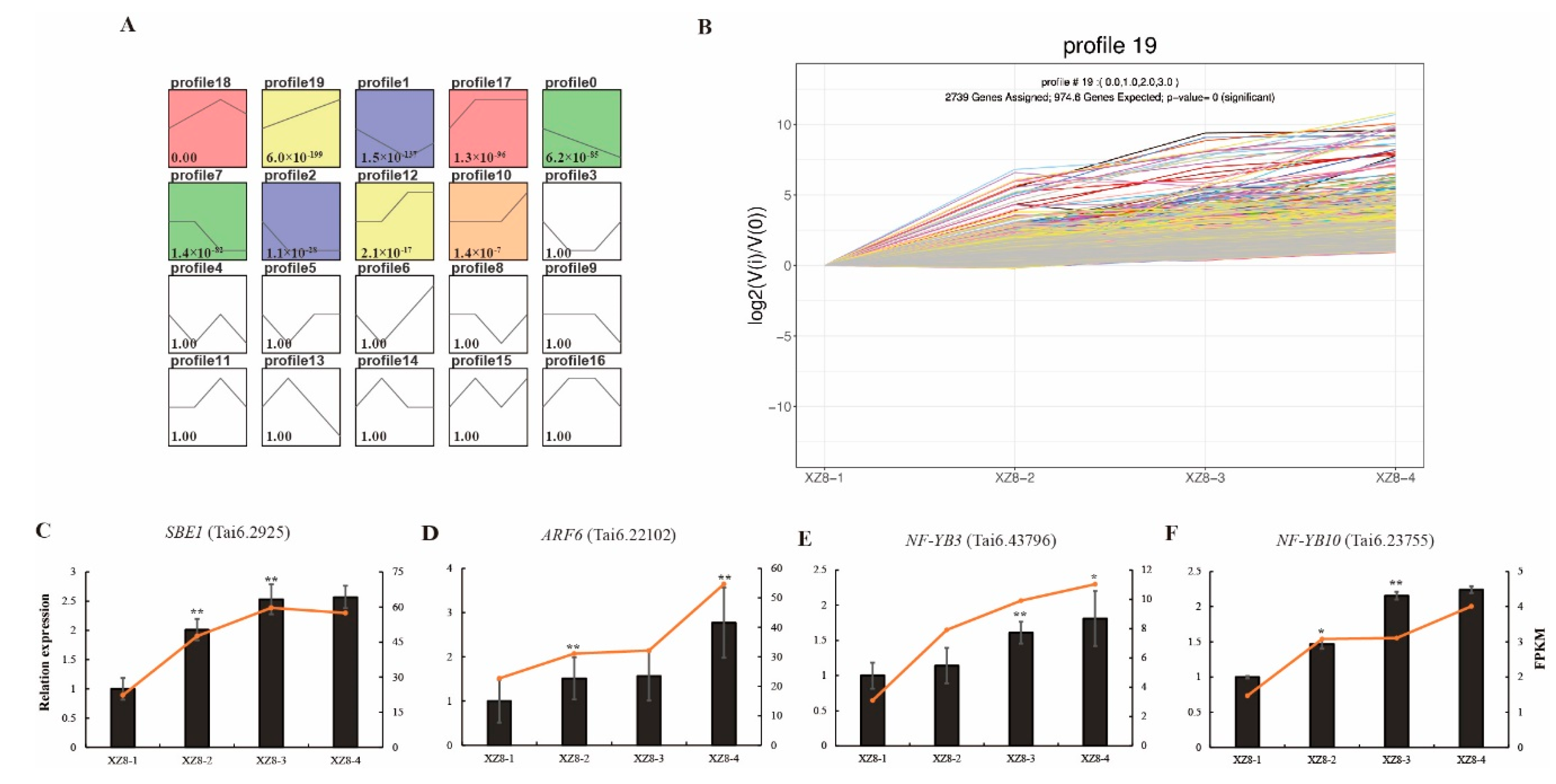
3.5. Validation of DEGs during SR Expansion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Fan, W.J.; Zhang, Y.D.; Wu, Y.L.; Zhou, W.Z.; Yang, J.; Yuan, L.; Zhang, P.; Wang, H.X. The H+-pyrophosphatase IbVP1 regulates carbon flux to influence the starch metabolism and yield of sweet potato. Hortic. Res. 2021, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Q.; Zhai, H.; Gao, S.P.; Yang, L.; Wang, Z.; Xu, Y.T.; Huo, J.X.; Ren, Z.T.; Zhao, N.; et al. IbBBX24 promotes the jasmonic acid pathway and enhances fusarium wilt resistance in sweet potato. Plant Cell 2020, 32, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Liu, Y.; Buitrago, S.; Jiang Wei Gao, H.R.; Han, H.; Wu, C.; Wang, Y.L.; Zhang, W.Y.; Yang, X.S. Adventitious root formation is dynamically regulated by various hormones in leaf-vegetable sweetpotato cuttings. J Plant Physiol. 2020, 253, 153267. [Google Scholar] [CrossRef]
- Zierer, W.; Rüscher, D.; Sonnewald, U.; Sonnewald, S. Tuber and tuberous root development. Annu. Rev. Plant Biol. 2021, 72, 551–580. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Z.; Li, X.; Gao, X.R.; Dai, Z.R.; Cui, Y.F.; Zhi, Y.H.; Liu, Q.C.; Zhai, H.; Gao, S.P.; et al. The IbBBX24-IbTOE3-IbPRX17 module enhances abiotic stress tolerance by scavenging reactive oxygen species in sweet potato. New Phytol. 2022, 233, 1133–1152. [Google Scholar] [CrossRef]
- Tang, C.; Han, R.P.; Zhou, Z.L.; Yang, Y.Y.; Zhu, M.K.; Xu, T.; Wang, A.M.; Li, Z.Y.; Dong, T.T. Identification of candidate miRNAs related in storage root development of sweet potato by high throughput sequencing. J. Plant Physiol. 2020, 251, 153224. [Google Scholar] [CrossRef]
- Duan, W.X.; Zhang, H.Y.; Xie, B.T.; Wang, B.Q.; Hou, F.Y.; Li, A.X.; Dong, S.X.; Qin, Z.; Wang, Q.M.; Zhang, L.M. Nitrogen utilization characteristics and early storage root development in nitrogen-tolerant and nitrogen-susceptible sweet potato. Physiol. Plant 2021, 173, 1090–1104. [Google Scholar] [CrossRef] [PubMed]
- Bararyenya, A.; Olukolu, B.A.; Tukamuhabwa, P.; Grüneberg, W.J.; Ekaya, W.; Low, J.; Ochwo-Ssemakula, M.; Odong, T.L.; Talwana, H.; Badji, A.; et al. Genome-wide association study identified candidate genes controlling continuous storage root formation and bulking in hexaploid sweetpotato. BMC Plant Biol. 2020, 20, 3. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Hamada, T.; Otani, M.; Shimada, T. Isolation and characterization of MADS box genes possibly related to root development in sweetpotato (Ipomoea batatas L. Lam.). J. Plant Biol. 2005, 48, 387–393. [Google Scholar] [CrossRef]
- Tanaka, M.; Takahata, Y.; Nakayama, H.; Nakatani, M.; Tahara, M. Altered carbohydrate metabolism in the storage roots of sweet potato plants overexpressing the SRF1 gene, which encodes a Dof zinc finger transcription factor. Planta 2009, 230, 737–746. [Google Scholar] [CrossRef]
- Noh, S.A.; Lee, H.S.; Huh, E.J.; Huh, G.M.; Paek, K.H.; Shin, J.S.; Bae, J.M. SRD1 is involved in the auxin-mediated initial thickening growth of storage root by enhancing proliferation of metaxylem and cambium cells in sweetpotato (Ipomoea batatas). J. Exp. Bot. 2010, 61, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.A.; Lee, H.S.; Kim, Y.S.; Paek, K.H.; Shin, J.S.; Bae, J.M. Down-regulation of the IbEXP1 gene enhanced storage root development in sweetpotato. J. Exp. Bot. 2013, 64, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.T.; Wang, H.X.; Hao, X.M.; Wu, Y.L.; Bian, X.F.; Yin, M.H.; Zhang, Y.D.; Fan, W.J.; Dai, H.; Yuan, L.; et al. Dynamic network biomarker analysis discovers IbNAC083 in the initiation and regulation of sweet potato root tuberization. Plant J. 2021, 108, 793–813. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.F.; Kim, H.S.; Kwak, S.S.; Zhang, Q.; Liu, S.; Ma, P.Y.; Jia, Z.D.; Xie, Y.Z.; Zhang, P.; Yu, Y. Different functions of IbRAP2.4, a drought-responsive AP2/ERF transcription factor, in regulating root development between Arabidopsis and sweetpotato. Front. Plant Sci. 2022, 13, 820450. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, X.B.; Zhao, L.K.; Huo, K.S.; Jin, P.F.; Zhao, D.L.; Zhou, Z.L.; Tang, J.; Xiao, S.Z.; Cao, Q.H. RNA-seq reveals the salt tolerance of Ipomoea pes-caprae, a wild relative of sweet potato. J. Plant Physiol. 2020, 255, 153276. [Google Scholar] [CrossRef]
- Li, Q.; Kou, M.; Li, C.; Zhang, Y.G. Comparative transcriptome analysis reveals candidate genes involved in anthocyanin biosynthesis in sweetpotato (Ipomoea batatas L.). Plant Physiol. Biochem. 2021, 158, 508–517. [Google Scholar] [CrossRef]
- Dong, T.; Zhu, M.; Yu, J.; Han, R.; Tang, C.; Xu, T.; Liu, J.; Li, Z. RNA-Seq and iTRAQ reveal multiple pathways involved in storage root formation and development in sweet potato (Ipomoea batatas L.). BMC Plant Biol. 2019, 19, 136. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Koestler, D.C.; Marsit, C.J.; Christensen, B.C.; Karagas, M.R.; Bueno, R.; Sugarbaker, D.J.; Kelsey, K.T.; Houseman, E.A. Semi-supervised recursively partitioned mixture models for identifying cancer subtypes. Bioinformatics 2010, 26, 2578–2585. [Google Scholar] [CrossRef]
- Skidmore, Z.L.; Wagner, A.H.; Lesurf, R.; Campbell, K.M.; Kunisaki, J.; Griffith, O.L.; Griffith, M. GenVisR: Genomic Visualizations in R. Bioinformatics 2016, 32, 3012–3014. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.R.; Chinnaiyan, A.M. Integrative analysis of the cancer transcriptome. Nat. Genet. 2005, 37, S31–S37. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.F.; Li, X.; Gao, S.P.; Nie, N.; Zhang, H.; Yang, Y.F.; He, S.Z.; Liu, Q.C.; Zhai, H. A novel WRKY transcription factor from Ipomoea trifida, ItfWRKY70, confers drought tolerance in sweet potato. Int. J. Mol. Sci. 2022, 23, 686. [Google Scholar] [CrossRef]
- Bahaji, A.; Li, J.; Sanchez-Lopeza, A.M.; Baroja-Fernández, E.; Muñoz, F.J.; Ovecka, M.; Almagro, G.; Montero, M.; Ezquer, I.; Etxeberria, E.; et al. Starch biosynthesis, its regulation and biotechnological approaches to improve crop yields. Biotechnol. Adv. 2014, 32, 87–106. [Google Scholar] [CrossRef]
- Kwak, M.S.; Noh, S.A.; Oh, M.J.; Huh, G.H.; Kim, K.N.; Lee, S.W.; Shin, J.S.; Bae, J.M. Two sweetpotato ADP-glucose pyrophosphorylase isoforms are regulated antagonistically in response to sucrose content in storage roots. Gene 2006, 366, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, S.T.; Xu, W.; Pu, Z.G.; Feng, J.Y.; Wang, Z.Y.; Zhang, C.; Peng, M.F.; Du, C.G.; Lin, F.; et al. The wild sweetpotato (Ipomoea trifida) genome provides insights into storage root development. BMC Plant Biol. 2019, 19, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.L.; He, S.Z.; Zhao, N.; Zhai, H.; Liu, Q.C. A sucrose non-fermenting-1-related protein kinase-1 gene, IbSnRK1, improves starch content, composition, granule size, degree of crystallinity and gelatinization in transgenic sweet potato. Plant Biotechnol. J. 2019, 17, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetlow, I.J.; Emes, M.J. A review of starch-branching enzymes and their role in amylopectin biosynthesis. IUBMB Life 2014, 66, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Hoang, N.V.; Park, C.; Kamran, M.; Lee, J.Y. Gene regulatory network guided investigations and engineering of storage root development in root crops. Front. Plant Sci. 2020, 11, 762. [Google Scholar] [CrossRef]
- Ponniah, S.K.; Thimmapuram, J.; Bhide, K.; Kalavacharla, V.K.; Manoharan, M. Comparative analysis of the root transcriptomes of cultivated sweetpotato (Ipomoea batatas (L.) Lam) and its wild ancestor (Ipomoea trifida [Kunth] G. Don). BMC Plant Biol. 2017, 17, 9. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Carmona, M.J.; Ortega, N.; Garcia-Maroto, F. Isolation and molecular characterization of a new vegetative MADS-box gene from Solanum tuberosum L. Planta 1998, 207, 181–188. [Google Scholar] [CrossRef]
- Rosin, F.M.; Hart, J.K.; Van Onckelen, H.; Hannapel, D.J. Suppression of a vegetative MADS box gene of potato activates axillary meristem development. Plant Physiol. 2003, 131, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Wang, Z.; Li, S.; Hou, M.; Zhou, Y.; Zhao, Y.; Li, G.; Zhao, H.; Ma, H. Genome-wide survey of potato MADS-box genes reveals that StMADS1 and StMADS13 are putative downstream targets of tuberigen StSP6A. BMC Genom. 2018, 19, 726. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Mizuno, K.; Fujimura, T. Isolation of MADS-box genes from sweet potato (Ipomoea batatas (L.) Lam.) expressed specifically in vegetative tissues. Plant Cell Physiol. 2002, 43, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, A.T.; Huang, Y.S.; Wang, Y.S.; Ma, D.F.; Yeh, K.W. IbMADS1 (Ipomoea batatasMADS-box 1 gene) is involved in tuberous root initiation in sweet potato (Ipomoea batatas). Ann. Bot. 2008, 102, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Rosin, F.M.; Prat, S.; Hannapel, D.J. Interacting transcription factors from the three-amino acid loop extension superclass regulate tuber formation. Plant Physiol. 2003, 132, 1391–1404. [Google Scholar] [CrossRef] [Green Version]
- Rosin, F.M.; Hart, J.K.; Horner, H.T.; Davies, P.J.; Hannapel, D.J. Overexpression of a knotted-like homeobox gene of potato alters vegetative development by decreasing gibberellin accumulation. Plant Physiol. 2003, 132, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Banerjee, A.K.; Hannapel, D.J. The tandem complex of BEL and KNOX partners is required for transcriptional repression of ga20ox1. Plant J. 2004, 38, 276–284. [Google Scholar] [CrossRef]
- Tanaka, M.; Kato, N.; Nakayama, H.; Nakatani, M.; Takahata, Y. Expression of class I knotted1-like homeobox genes in the storage roots of sweetpotato (Ipomoea batatas). J. Plant Physiol. 2008, 165, 1726–1735. [Google Scholar] [CrossRef]
- Guilfoyle, T.J.; Hagen, G. Auxin response factors. Curr. Opin. Plant Biol. 2007, 10, 453–460. [Google Scholar] [CrossRef]
- Petroni, K.; Kumimoto, R.W.; Gnesutta, N.; Calvenzani, V.; Fornari, M.; Tonelli, C.; Holt, B.F., 3rd; Mantovani, R. The promiscuous life of plant NUCLEAR FACTOR Y transcription factors. Plant Cell 2012, 24, 4777–4792. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, W.; Yan, H.; Ma, M.; Kou, M.; Li, C.; Tang, W.; Yu, Y.; Hao, Q.; Nguyen, T.; Wang, X.; et al. Comparative Transcriptome Profiling Reveals the Genes Involved in Storage Root Expansion in Sweetpotato (Ipomoea batatas (L.) Lam.). Genes 2022, 13, 1156. https://doi.org/10.3390/genes13071156
Song W, Yan H, Ma M, Kou M, Li C, Tang W, Yu Y, Hao Q, Nguyen T, Wang X, et al. Comparative Transcriptome Profiling Reveals the Genes Involved in Storage Root Expansion in Sweetpotato (Ipomoea batatas (L.) Lam.). Genes. 2022; 13(7):1156. https://doi.org/10.3390/genes13071156
Chicago/Turabian StyleSong, Weihan, Hui Yan, Meng Ma, Meng Kou, Chen Li, Wei Tang, Yicheng Yu, Qixian Hao, Thanhliem Nguyen, Xin Wang, and et al. 2022. "Comparative Transcriptome Profiling Reveals the Genes Involved in Storage Root Expansion in Sweetpotato (Ipomoea batatas (L.) Lam.)" Genes 13, no. 7: 1156. https://doi.org/10.3390/genes13071156
APA StyleSong, W., Yan, H., Ma, M., Kou, M., Li, C., Tang, W., Yu, Y., Hao, Q., Nguyen, T., Wang, X., Zhang, Z., You, C., Gao, R., Zhang, Y., & Li, Q. (2022). Comparative Transcriptome Profiling Reveals the Genes Involved in Storage Root Expansion in Sweetpotato (Ipomoea batatas (L.) Lam.). Genes, 13(7), 1156. https://doi.org/10.3390/genes13071156