
Genome-Wide Evolutionary Analysis of Putative Non-Specific Herbicide Resistance Genes and Compilation of Core Promoters between Monocots and Dicots



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval
2.2. Identification of UDPGT, GST, NMO, and CytochromeP450
2.3. Phylogenetic Study
2.4. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Pathway Analysis
2.5. In Silico Expression Analysis of NTSR Genes
2.6. Identification of Cis Motifs and Transcription Factors Binding Sites in Promoters of NTSR Genes
2.7. Homology Modelling and Protein Secondary Structure Assignment
3. Results
3.1. Identification of Putative NTSR Genes
3.2. Gene Ontology and KEGG Pathway Analysis
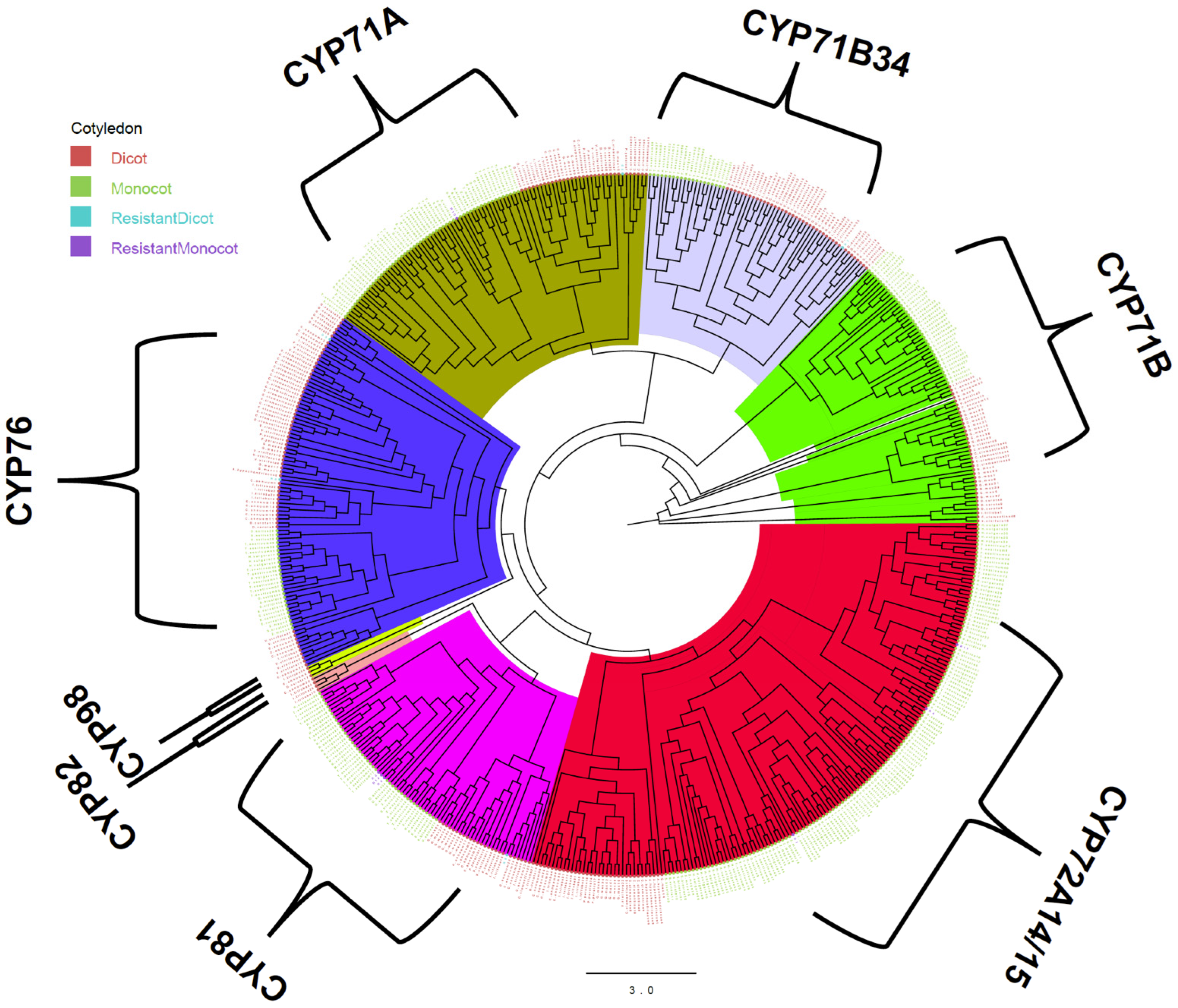
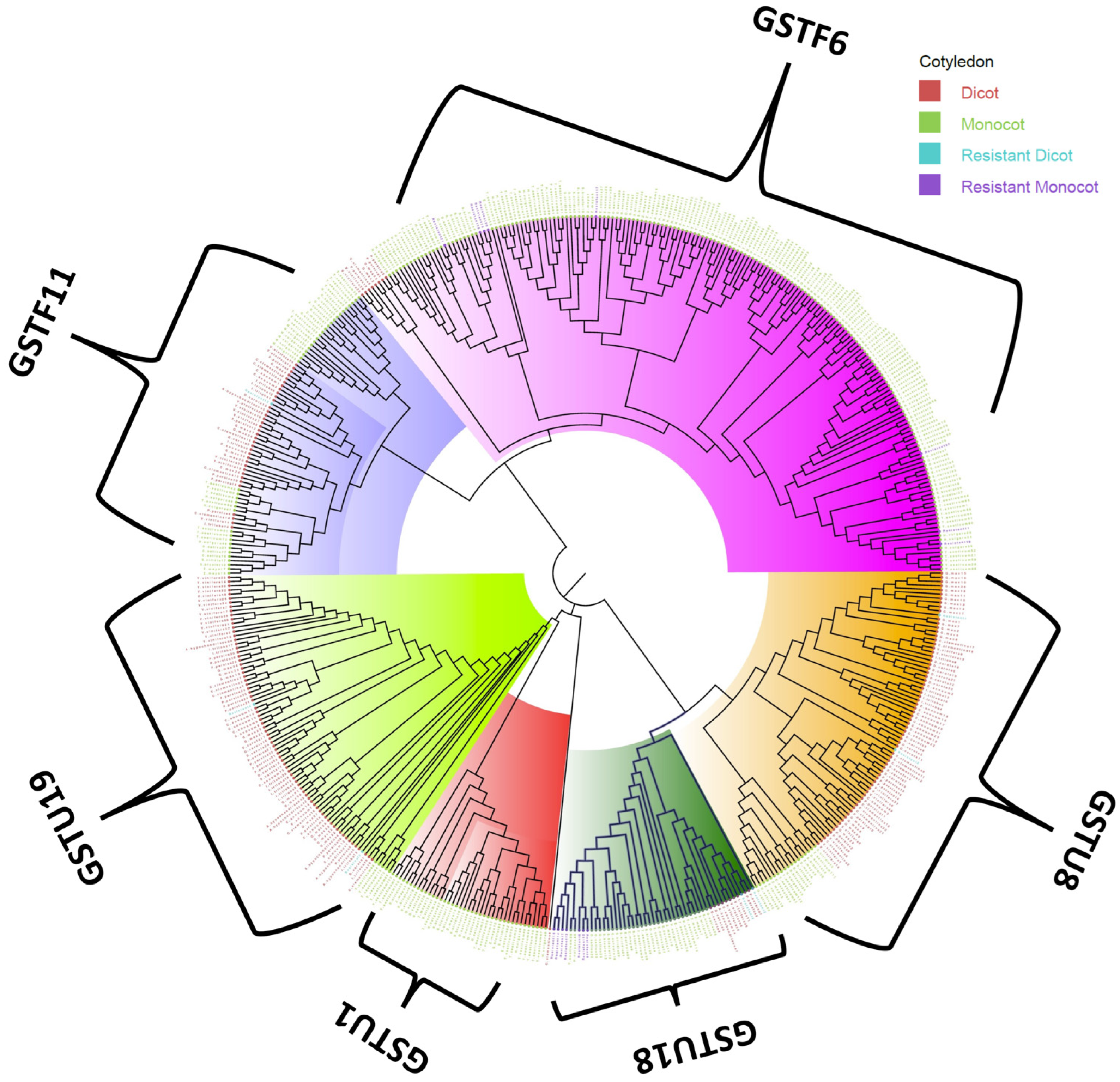
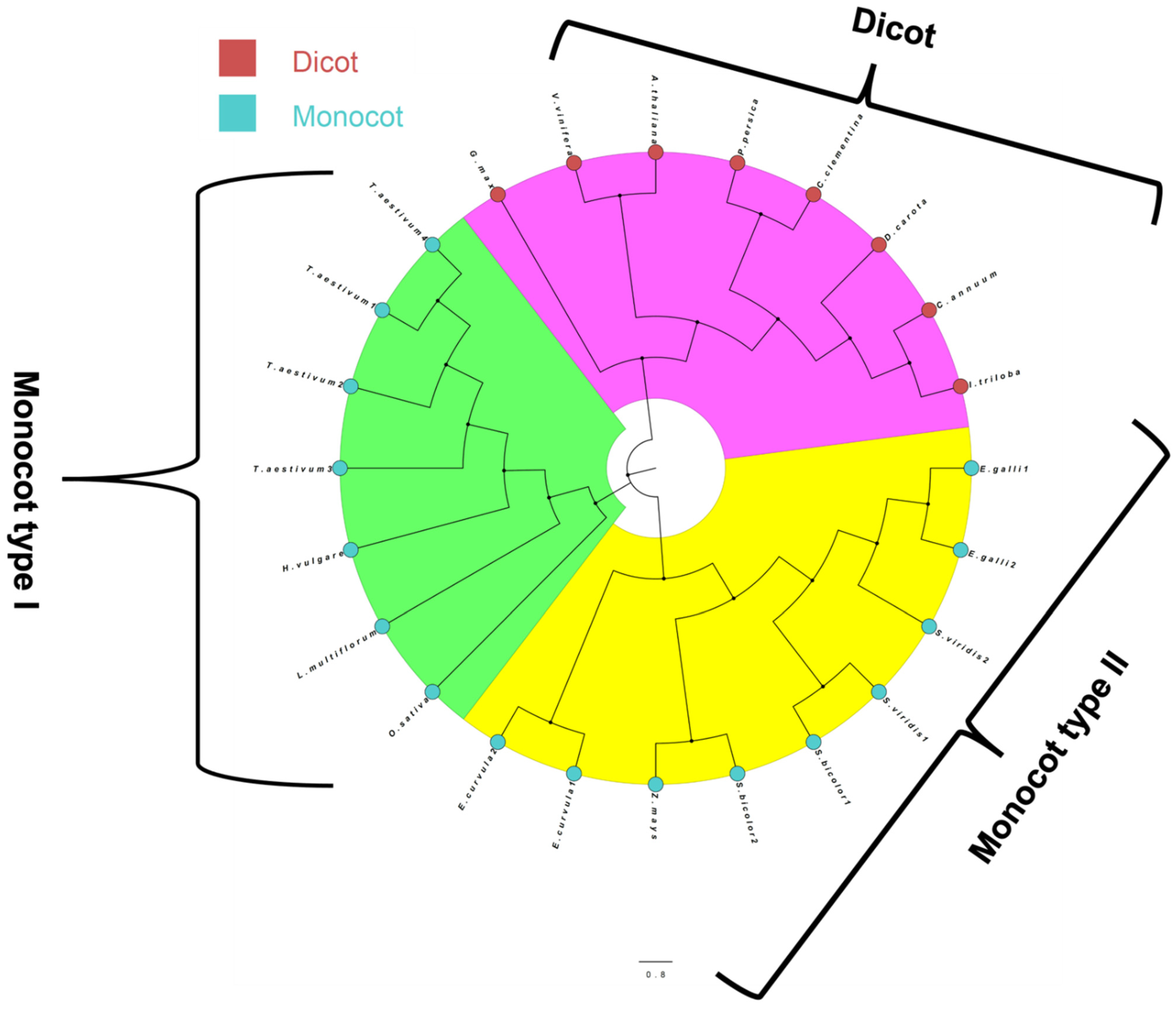
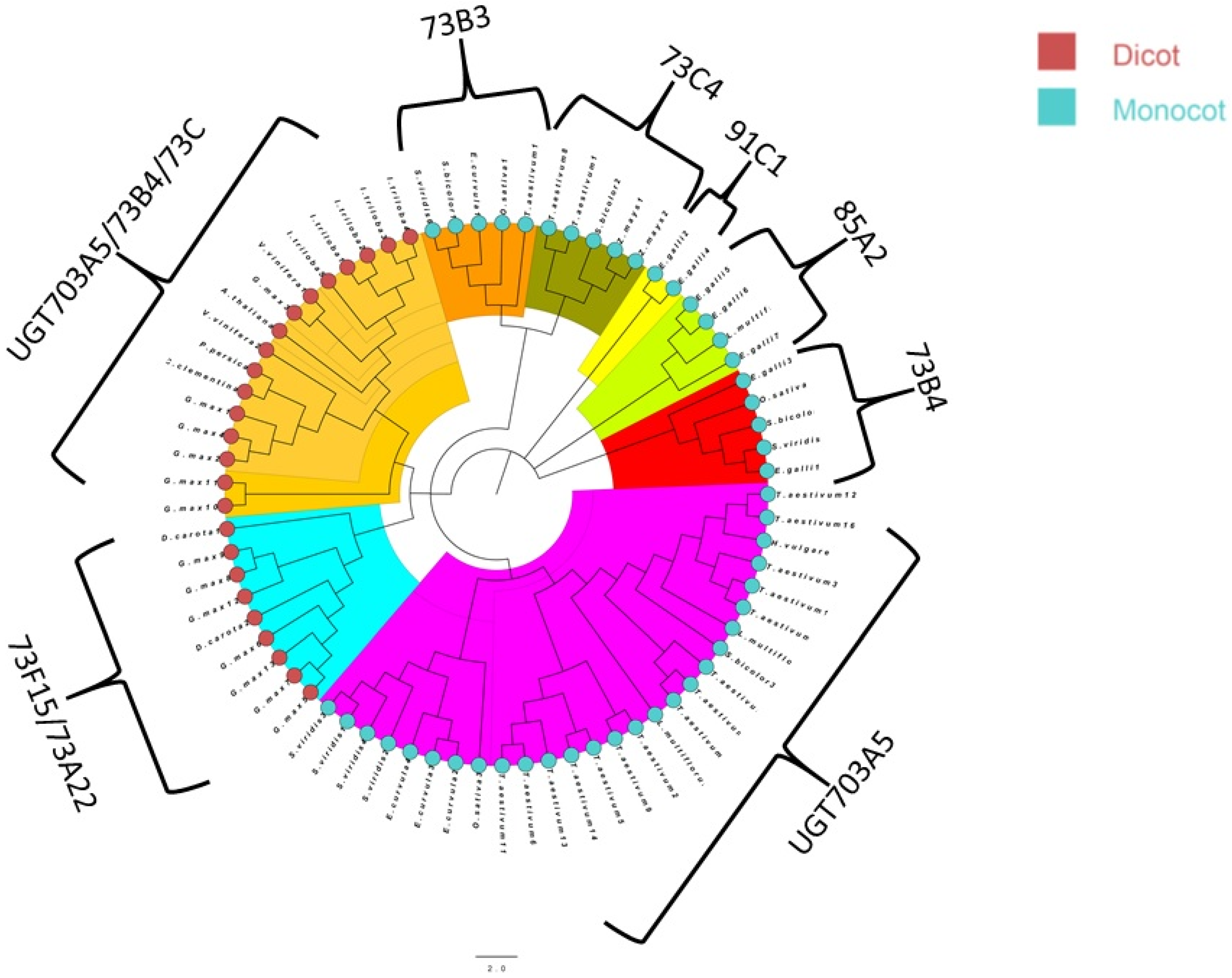
3.3. Phylogenetic Analysis
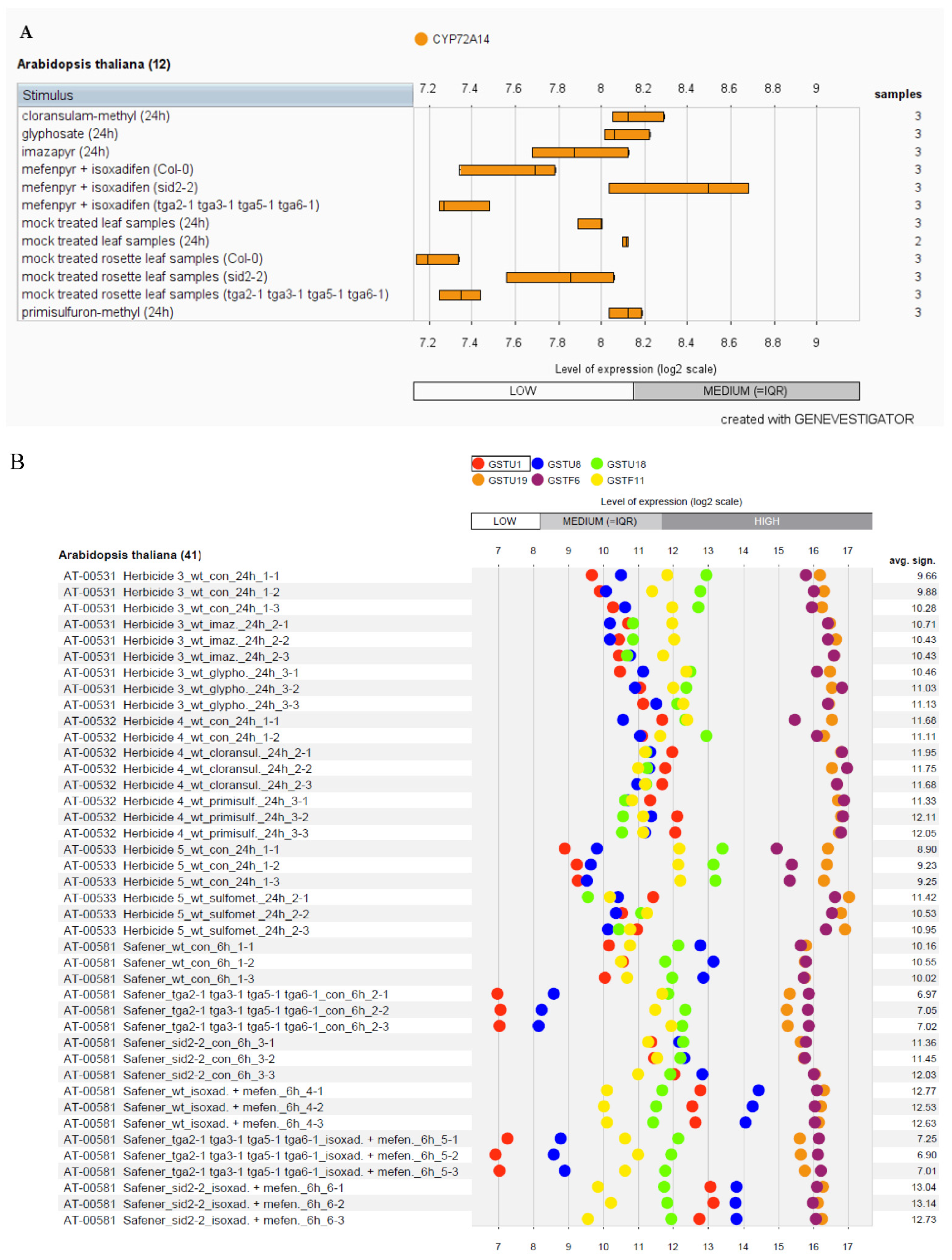
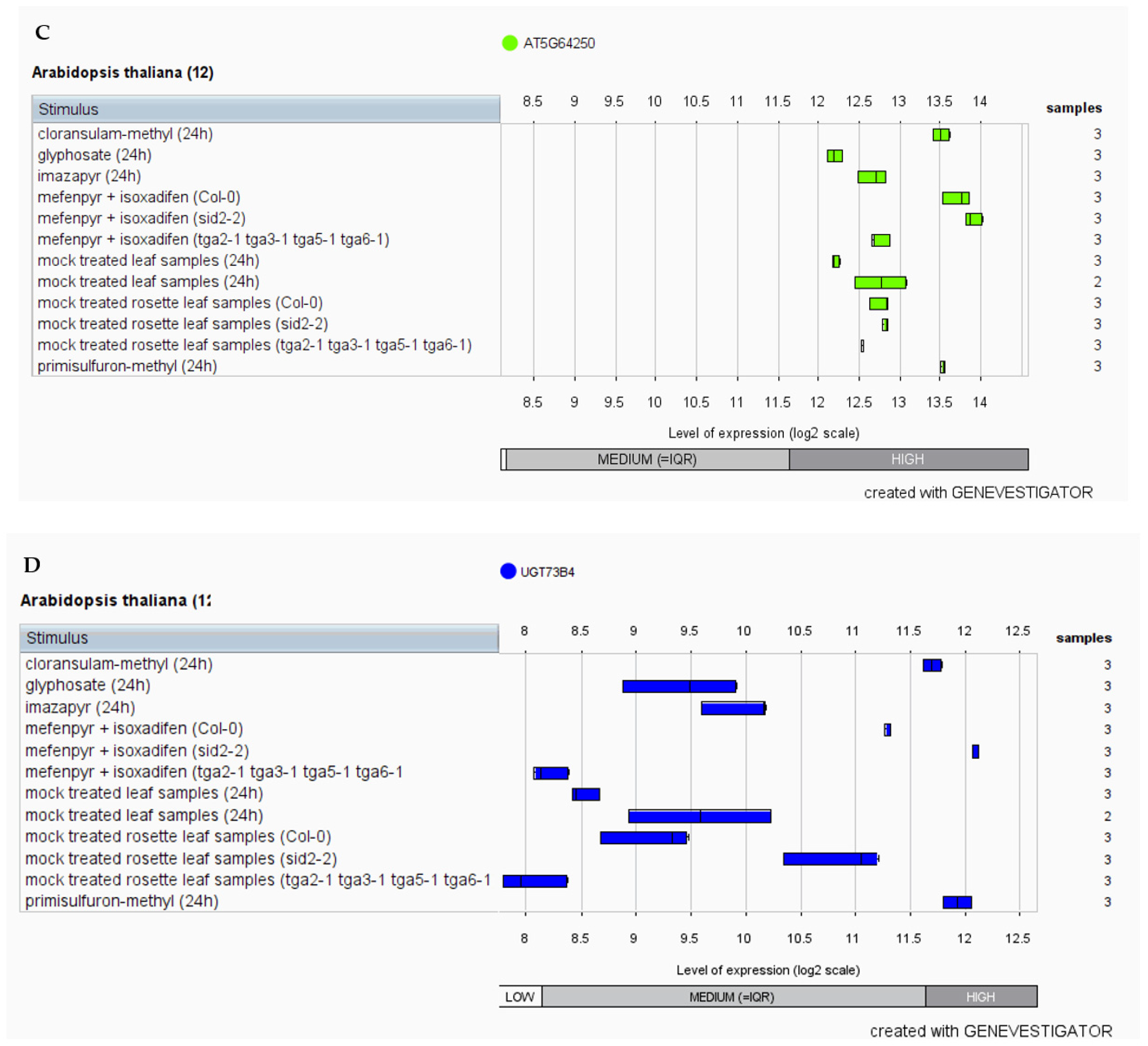
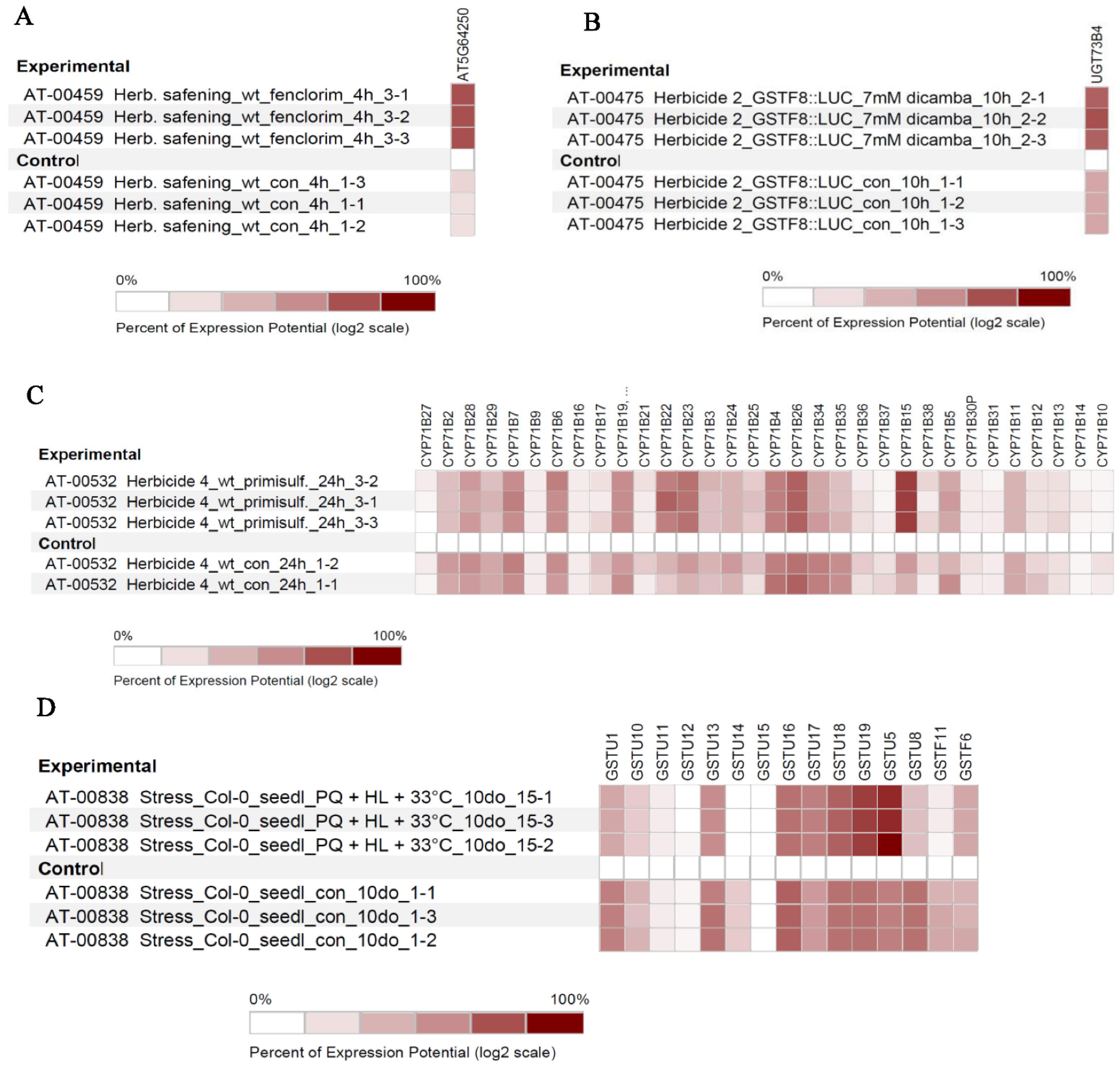
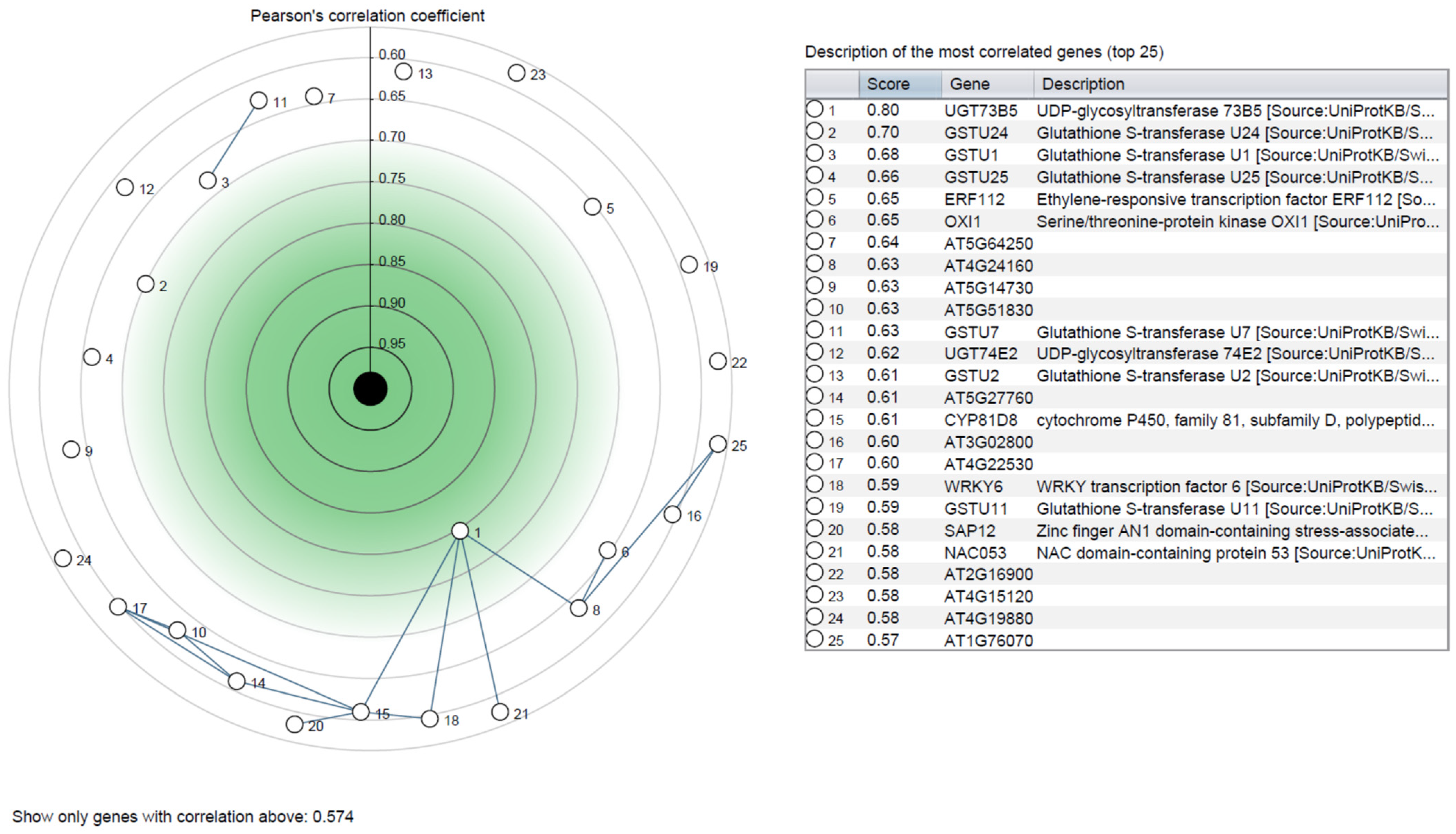
3.4. In-Silico Expression Analysis
3.5. Identification of Cis-Motifs and TFBS in Promoters
3.6. Homology Modelling of Resistant CYP450 and GST Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Switzer, C.M.; Whitehead, C.W. The differential response of strains of wild carrot to 2,4-D and related herbicides. Can. J. Plant Sci. 1963, 43, 255–262. [Google Scholar]
- Peterson, M.A.; Collavo, A.; Ovejero, R.; Shivrain, V.; Walsh, M.J. The challenge of herbicide resistance around the world: A current summary. Pest Manag. Sci. 2018, 74, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Heap, I. International Survey of Herbicide Resistant Weeds. Available online: https://www.weedscience.org (accessed on 30 June 2021).
- Kaundun, S.S. Syngenta’s contribution to herbicide resistance research and management. Pest Manag. Sci. 2021, 77, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Powles, S.B.; Yu, Q. Evolution in action: Plants resistant to herbicides. Annu. Rev. Plant Biol. 2010, 61, 317–347. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Yu, Q.; Beffa, R.; González, S.; Maiwald, F.; Wang, J.; Powles, S.B. Cytochrome P450 CYP81A10v7 in Lolium rigidum confers metabolic resistance to herbicides across at least five modes of action. Plant J. 2021, 105, 79–92. [Google Scholar] [CrossRef]
- Perotti, V.E.; Larran, A.S.; Palmieri, V.E.; Martinatto, A.K.; Permingeat, H.R. Herbicide resistant weeds: A call to integrate conventional agricultural practices, molecular biology knowledge and new technologies. Plant Sci. 2020, 290, 110255. [Google Scholar] [CrossRef]
- Gaines, T.A.; Duke, S.O.; Morran, S.; Rigon, C.A.; Tranel, P.J.; Kupper, A.; Dayan, F.E. Mechanisms of evolved herbicide resistance. J. Biol. Chem. 2020, 295, 10307–10330. [Google Scholar] [CrossRef]
- Mahmood, K.; Mathiassen, S.K.; Kristensen, M.; Kudsk, P. Multiple herbicide resistance in Lolium multiflorum and identification of conserved regulatory elements of herbicide resistance genes. Front. Plant Sci. 2016, 7, 1160. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Yu, Q.; Han, H.; Mao, L.; Nyporko, A.; Fan, L.; Bai, L.; Powles, S. Aldo-keto reductase metabolizes glyphosate and confers glyphosate resistance in Echinochloa colona. Plant Physiol. 2019, 181, 1519–1534. [Google Scholar] [CrossRef]
- Yu, Q.; Powles, S. Metabolism-based herbicide resistance and cross-resistance in crop weeds: A threat to herbicide sustainability and global crop production. Plant Physiol. 2014, 166, 1106–1118. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Deng, W.; Li, X.; Yu, Q.; Bai, L.; Zheng, M. Target-site and non-target-site based resistance to the herbicide tribenuron-methyl in flixweed (Descurainia sophia L.). BMC Genom. 2016, 17, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busi, R.; Porri, A.; Gaines, T.A.; Powles, S.B. Pyroxasulfone resistance in Lolium rigidum is metabolism-based. Pestic. Biochem. Physiol. 2018, 148, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Iwakami, S.; Kamidate, Y.; Yamaguchi, T.; Ishizaka, M.; Endo, M.; Suda, H.; Nagai, K.; Sunohara, Y.; Toki, S.; Uchino, A.; et al. CYP 81A P450s are involved in concomitant cross-resistance to acetolactate synthase and acetyl-CoA carboxylase herbicides in Echinochloa phyllopogon. New Phytol. 2019, 221, 2112–2122. [Google Scholar] [CrossRef]
- Ma, R.; Kaundun, S.S.; Tranel, P.J.; Riggins, C.W.; McGinness, D.L.; Hager, A.G.; Hawkes, T.; McIndoe, E.; Riechers, D.E. Distinct detoxification mechanisms confer resistance to mesotrione and atrazine in a population of waterhemp. Plant Physiol. 2013, 163, 363–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duke, S.O. Why have no new herbicide modes of action appeared in recent years? Pest Manag. Sci. 2012, 68, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittenbach, V.A.; Abell, L.M. Inhibitors of valine, leucine, and isoleucine biosynthesis. In Plant Amino Acids; Singh, B.K., Ed.; Marcel Dekker: New York, NY, USA, 1999; pp. 385–416. [Google Scholar]
- Childers, A. Sequenced Arthropod Genomes, i5k Initiative. 2018. Available online: http://i5k.github.io/arthropod_genomes_at_ncbi#FAQ (accessed on 5 November 2021).
- i5K Consortium. The i5K Initiative: Advancing arthropod genomics for knowledge, human health, agriculture, and the environment. J. Hered. 2013, 104, 595–600. [Google Scholar] [CrossRef]
- Pedro, H.; Maheswari, U.; Urban, M.; Irvine, A.G.; Cuzick, A.; McDowall, M.D.; Staines, D.M.; Kulesha, E.; Hammond-Kosack, K.E.; Kersey, P.J. PhytoPath: An integrative resource for plant pathogen genomics. Nucleic Acids Res. 2016, 44, D688–D693. [Google Scholar] [CrossRef] [Green Version]
- Clouse, J.W.; Adhikary, D.; Page, J.T.; Ramaraj, T.; Deyholos, M.K.; Udall, J.A.; Fairbanks, D.J.; Jellen, E.N.; Maughan, P.J. The amaranth genome: Genome, transcriptome, and physical map assembly. Plant Genome 2016, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dorn, K.M.; Fankhauser, J.D.; Wyse, D.L.; Marks, M.D. A draft genome of field pennycress (Thlaspi arvense) provides tools for the domestication of a new winter biofuel crop. DNA Res. 2015, 22, 121–131. [Google Scholar] [CrossRef]
- Guo, L.; Qiu, J.; Ye, C.; Jin, G.; Mao, L.; Zhang, H.; Yang, X.; Peng, Q.; Wang, Y.; Longjiang, F.; et al. Echinochloa crus-galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nat. Commun. 2017, 8, 1031. [Google Scholar] [CrossRef]
- Moghe, G.D.; Hufnagel, D.E.; Tang, H.; Xiao, Y.; Dworkin, I.; Town, C.D.; Conner, J.K.; Shiu, S.H. Consequences of whole-genome triplication as revealed by comparative genomic analyses of the wild radish Raphanus raphanistrum and three other Brassicaceae species. Plant Cell 2014, 26, 1925–1937. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, J.S.; Giacomini, D.; Waithaka, B.; Lanz, C.; Murphy, B.P.; Campe, R.; Lerchl, J.; Landes, A.; Gatzmann, F.; Janssen, A.; et al. Draft Genomes of Amaranthus tuberculatus, Amaranthus hybridus, and Amaranthus palmeri. Genome Biol. Evol. 2020, 12, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Lai, Z.; Lane, T.; Nageswara-Rao, M.; Okada, M.; Jasieniuk, M.; O’Geen, H.; Kim, R.W.; Sammons, R.D.; Rieseberg, L.H.; et al. De novo genome assembly of the economically important weed horseweed using integrated data from multiple sequencing platforms. Plant Physiol. 2014, 166, 1241–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravet, K.; Patterson, E.; Krähmer, H.; Hamouzova, K.; Fan, L.; Jasieniuk, M.; Lawton-Rauh, A.; Malone, J.M.; McElroy, J.S.; Merotto, A.; et al. The power and potential of genomics in weed biology and management. Pest Manag. Sci. 2018, 74, 2216–2225. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.M.; Arsovski, A.A.; Lempe, J.; Bubb, K.L.; Weirauch, M.T.; Sabo, P.J.; Sandstrom, R.; Thurman, R.E.; Neph, S.; Reynolds, A.P.; et al. Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Rep. 2014, 8, 2015–2030. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Assmann, S.M.; Albert, R. Predicting essential components of signal transduction networks: A dynamic model of guard cell abscisic acid signaling. PLoS Biol. 2006, 4, e312. [Google Scholar] [CrossRef]
- Muraro, D.; Simmons, A. An integrative analysis of gene expression and molecular interaction data to identify dys-regulated sub-networks in inflammatory bowel disease. BMC Bioinform. 2016, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Forrest, A.R.R.; Nimwegen, E.V.; Daub, C.; Balwierz, P.J.; Irvine, K.M.; Lassmann, T.; Ravasi, T.; Hasegawa, Y.; de Hoon, M.J.L.; et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat. Genet. 2009, 41, 553–562. [Google Scholar]
- Knorst, V.; Yates, S.; Byrne, S.; Asp, T.; Widmer, F.; Studer, B.; Kolliker, R. First assembly of the gene-space of Lolium multiflorum and comparison to other Poaceae genomes. Grassl. Sci. 2019, 65, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tossato, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39 (Suppl. 2), W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Dueholm, B.; Krieger, C.; Drew, D.; Olry, A.; Kamo, T.; Taboureau, O.; Weitzel, C.; Bourgaud, F.; Hehn, A.; Simonsen, H.T. Evolution of substrate recognition sites (SRSs) in cytochromes P450 from Apiaceae exemplified by the CYP71AJ subfamily. BMC Evol. Biol. 2015, 15, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Behringer, C.; Bartsch, K.; Schaller, A. Safeners recruit multiple signalling pathways for the orchestrated induction of the cellular xenobiotic detoxification machinery in Arabidopsis. Plant Cell Environ. 2011, 34, 1970–1985. [Google Scholar] [CrossRef]
- Das, M.; Reichman, J.R.; Haberer, G.; Welzl, G.; Aceituno, F.F.; Mader, M.T.; Watrud, L.S.; Pfleeger, T.G.; Gutierrez, R.A.; Schaffner, A.R.; et al. A composite transcriptional signature differentiates responses towards closely related herbicides in Arabidopsis thaliana and Brassica napus. Plant Mol. Biol. 2010, 72, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Gleason, C.; Foley, R.C.; Singh, K.B. Mutant analysis in Arabidopsis provides insight into the molecular mode of action of the auxinic herbicide dicamba. PLoS ONE 2011, 6, e17245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skipsey, M.; Knight, K.M.; Brazier-Hicks, M.; Dixon, D.P.; Steel, P.G.; Edwards, R. Xenobiotic responsiveness of Arabidopsis thaliana to a chemical series derived from a herbicide safener. J. Biol. Chem. 2011, 286, 32268–32276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandalinas, S.I.; Sengupta, S.; Fritschi, F.B.; Azad, R.K.; Nechushtai, R.; Mittler, R. The impact of multifactorial stress combination on plant growth and survival. New Phytol. 2021, 230, 1034–1048. [Google Scholar] [CrossRef] [PubMed]
- Délye, C. Unravelling the genetic bases of non-target-site-based resistance (NTSR) to herbicides: A major challenge for weed science in the forthcoming decade. Pest Manag. Sci. 2013, 69, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Chavira, I.; Contreras-Vergara, C.A.; Arvizu-Flores, A.A.; Serrano-Posada, H.; Lopez-Zavala, A.A.; García-Orozco, K.D.; Hernandez-Paredes, J.; Rudiño-Piñera, E.; Stojanoff, V.; Sotelo Mundo, R.R.; et al. Insights into ligand binding to a glutathione S-transferase from mango: Structure, thermodynamics and kinetics. Biochimie 2017, 135, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Kreiner, J.M.; Giacomini, D.A.; Bemm, F.; Waithaka, B.; Regalado, J.; Lanz, C.; Hildebrandt, J.; Sikkema, P.H.; Tranel, P.J.; Weigel, D.; et al. Multiple modes of convergent adaptation in the spread of glyphosate-resistant Amaranthus tuberculatus. Proc. Natl. Acad. Sci. USA 2019, 116, 21076–21084. [Google Scholar] [CrossRef] [Green Version]
- Neve, P.; Busi, R.; Renton, M.; Vila-Aiub, M.M. Expanding the eco-evolutionary context of herbicide resistance research. Pest Manag. Sci. 2014, 70, 1385–1393. [Google Scholar] [CrossRef]
- Nelson, D.; Werck-Reichhart, D. A P450-centric view of plant evolution. Plant J. 2011, 66, 194–211. [Google Scholar] [CrossRef]
- Gaines, T.A.; Lorentz, L.; Figge, A.; Herrmann, J.; Maiwald, F.; Ott, M.C.; Han, H.; Busi, R.; Yu, Q.; Powles, S.B.; et al. RNA-Seq transcriptome analysis to identify genes involved in metabolism-based diclofop resistance in Lolium rigidum. Plant J. 2014, 78, 865–876. [Google Scholar] [CrossRef]
- Hatton, P.J.; Dixon, D.; Cole, D.J.; Edwards, R. Glutathione transferase activities and herbicide selectivity in maize and associated weed species. Pestic. Sci. 1996, 46, 267–275. [Google Scholar] [CrossRef]
- Cummins, I.; Dixon, D.P.; Freitag-Pohl, S.; Skipsey, M.; Edwards, R. Multiple roles for plant glutathione transferases in xenobiotic detoxification. Drug Metab. Rev. 2011, 43, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, N.; Poudel, N.; Vlachakis, D.; Papageorgiou, A.C.; Labrou, N.E. Phi class glutathione transferases as molecular targets towards multiple-herbicide resistance: Inhibition analysis and pharmacophore design. Plant Physiol. Biochem. 2021, 158, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Dimaano, N.G.; Iwakami, S. Cytochrome P450-mediated herbicide metabolism in plants: Current understanding and prospects. Pest Manag. Sci. 2021, 77, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Siminszky, B. Plant cytochrome P450-mediated herbicide metabolism. Phytochem. Rev. 2006, 5, 445–458. [Google Scholar] [CrossRef]
- Yanniccari, M.; Gigón, R.; Larsen, A. Cytochrome P450 herbicide metabolism as the main mechanism of cross-resistance to ACCase-and ALS-inhibitors in Lolium spp. populations from Argentina: A molecular approach in characterization and detection. Front. Plant Sci. 2020, 11, 600301. [Google Scholar] [CrossRef]
- Salas-Perez, R.A.; Saski, C.A.; Noorai, R.E.; Srivastava, S.K.; Lawton-Rauh, A.L.; Nichols, R.L.; Roma-Burgos, N. RNA-Seq transcriptome analysis of Amaranthus palmeri with differential tolerance to glufosinate herbicide. PLoS ONE 2018, 13, e0195488. [Google Scholar] [CrossRef]
- Werck-Reichhart, D.; Hehn, A.; Didierjean, L. Cytochromes P450 for engineering herbicide tolerance. Trends Plant Sci. 2000, 5, 116–123. [Google Scholar] [CrossRef]
- Pan, L.; Gao, H.; Xia, W.; Zhang, T.; Dong, L. Establishing a herbicide-metabolizing enzyme library in Beckmannia syzigachne to identify genes associated with metabolic resistance. J. Exp. Bot. 2016, 67, 1745–1757. [Google Scholar] [CrossRef]
- Vega, T.; Gil, M.; Martin, G.; Moschen, S.; Picardi, L.; Nestares, G. Stress response and detoxification mechanisms involved in non-target-site herbicide resistance in sunflower. Crop Sci. 2020, 60, 1809–1822. [Google Scholar] [CrossRef]
- Bowles, D.; Isayenkova, J.; Lim, E.K.; Poppenberger, B. Glycosyltransferases: Managers of small molecules. Curr. Opin. Plant Biol. 2005, 8, 254–263. [Google Scholar] [CrossRef]
- Gandia-Herrero, F.; Lorenz, A.; Larson, T.; Graham, I.A.; Bowles, D.J.; Rylott, E.L.; Bruce, N.C. Detoxification of the explosive 2, 4, 6-trinitrotoluene in Arabidopsis: Discovery of bifunctional O-and C-glucosyltransferases. Plant J. 2008, 56, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Matzrafi, M.; Shaar-Moshe, L.; Rubin, B.; Peleg, Z. Unraveling the transcriptional basis of temperature-dependent pinoxaden resistance in Brachypodium hybridum. Front. Plant Sci. 2017, 8, 1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigon, C.A.; Gaines, T.A.; Küpper, A.; Dayan, F.E. Metabolism-based herbicide resistance, the major threat among the non-target site resistance mechanisms. Outlooks Pest Manag. 2020, 31, 162–168. [Google Scholar] [CrossRef]
- Chen, X.; Xia, J.; Shang, Q.; Song, D.; Gao, X. UDP-glucosyltransferases potentially contribute to imidacloprid resistance in Aphis gossypii glover based on transcriptomic and proteomic analyses. Pestic. Biochem. Physiol. 2019, 159, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Brazier, M.; Cole, D.J.; Edwards, R. O-glucosyltransferase activities toward phenolic natural products and xenobiotics in wheat and herbicide-resistant and herbicide-susceptible black-grass (Alopecurus myosuroides). Phytochemistry 2002, 59, 149–156. [Google Scholar] [CrossRef]
- Nagpal, A.; Valley, M.P.; Fitzpatrick, P.F.; Orville, A.M. Crystal structures of nitroalkane oxidase: Insights into the reaction mechanism from a covalent complex of the flavoenzyme trapped during turnover. Biochemistry 2006, 45, 1138–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadda, G.; Francis, K. Nitronate monooxygenase, a model for anionic flavin semiquinone intermediates in oxidative catalysis. Arch. Biochem. Biophys. 2010, 493, 53–61. [Google Scholar] [CrossRef]
- Torres-Guzman, J.C.; Padilla-Guerrero, I.E.; Cervantes-Quintero, K.Y.; Martinez-Vazquez, A.; Ibarra-Guzman, M.; Gonzalez-Hernandez, G.A. Peculiarities of nitronate monooxygenases and perspectives for in vivo and in vitro applications. Appl. Microbiol. Biotechnol. 2021, 105, 8019–8032. [Google Scholar] [CrossRef]
- Jensen, K.; Møller, B.L. Plant NADPH-cytochrome P450 oxidoreductases. Phytochemistry 2010, 71, 132–141. [Google Scholar] [CrossRef]
- Li, Y.; Wei, K. Comparative functional genomics analysis of cytochrome P450 gene superfamily in wheat and maize. BMC Plant Biol. 2020, 20, 93. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Sahoo, A.; Devendran, R.; Jain, M. Over-expression of a rice tau class glutathione s-transferase gene improves tolerance to salinity and oxidative stresses in Arabidopsis. PLoS ONE 2014, 9, e92900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, B.; Lopes, J.; Leal, A.; Martins, M.; Soares, C.; Azenha, M.; Fidalgo, F.; Teixeira, J. Specific glutathione-S-transferases ensure an efficient detoxification of diclofenac in Solanum lycopersicum L. plants. Plant Physiol. Biochem. 2021, 168, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Iwakami, S.; Endo, M.; Saika, H.; Okuno, J.; Nakamura, N.; Yokoyama, M.; Watanabe, H.; Toki, S.; Uchino, A.; Inamura, T. Cytochrome P450 CYP81A12 and CYP81A21 are associated with resistance to two acetolactate synthase inhibitors in Echinochloa phyllopogon. Plant Physiol. 2014, 165, 618–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Ortega, S.; Goldberg-Cavalleri, A.; Walker, A.; Brazier-Hicks, M.; Onkokesung, N.; Edwards, R. Non-target site herbicide resistance is conferred by two distinct mechanisms in black-grass (Alopecurus myosuroides). Front. Plant Sci. 2021, 12, 194. [Google Scholar] [CrossRef]
- Benedetti, L.; Rangani, G.; Ebeling-Viana, V.; Carvalho-Moore, P.; Rabaioli-Camargo, E.; de Avila, L.A.; Roma-Burgos, N. Recurrent selection by herbicide sublethal dose and drought stress results in rapid reduction of herbicide sensitivity in junglerice. Agronomy 2020, 10, 1619. [Google Scholar] [CrossRef]
- Rouse, C.E. Characterization of Multiple-Herbicide-Resistant Echinochloa colona from Arkansas. Ph.D. Dissertation, University of Arkansas, Fayetteville, AR, USA, December 2017. [Google Scholar]
- Gallé, Á.; Czékus, Z.; Bela, K.; Horváth, E.; Ördög, A.; Csiszár, J.; Poór, P. Plant glutathione transferases and light. Front. Plant Sci. 2019, 9, 1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangani, G.; Noguera, M.; Salas-Perez, R.; Benedetti, L.; Roma-Burgos, N. Mechanism of resistance to S-metolachlor in Palmer amaranth. Front. Plant Sci. 2021, 12, 652581. [Google Scholar] [CrossRef]
- Tétard-Jones, C.; Sabbadin, F.; Moss, S.; Hull, R.; Neve, P.; Edwards, R. Changes in the proteome of the problem weed blackgrass correlating with multiple-herbicide resistance. Plant J. 2018, 94, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Yonekura-Sakakibara, K.; Hanada, K. An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J. 2011, 66, 182–193. [Google Scholar] [CrossRef]
- Ouyang, D.; Wang, L.C.; Tang, T.; Feng, H. Genomic-wide identification and characterization of the Uridine Diphosphate Glycosyltransferase Family in Eucommia ulmoides Oliver. Plants 2021, 10, 1934. [Google Scholar] [CrossRef]
- Karavangeli, M.; Labrou, N.E.; Clonis, Y.D.; Tsaftaris, A. Development of transgenic tobacco plants overexpressing maize glutathione S-transferase I for chloroacetanilide herbicides phytoremediation. Biomol. Eng. 2005, 22, 121–128. [Google Scholar] [CrossRef]
- Liu, X.; Xu, X.; Li, B.; Wang, X.; Wang, G.; Li, M. RNA-seq transcriptome analysis of maize inbred carrying nicosulfuron-tolerant and nicosulfuron-susceptible alleles. Int. J. Mol. Sci. 2015, 16, 5975–5989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanhere, A.; Bansal, M. Structural properties of promoters: Similarities and differences between prokaryotes and eukaryotes. Nucleic Acids Res. 2005, 33, 3165–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietz, K.J.; Vogel, M.O.; Viehhauser, A. AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signalling. Protoplasma 2010, 245, 3–14. [Google Scholar] [CrossRef]
- Rabara, R.C.; Tripathi, P.; Rushton, P.J. The potential of transcription factor-based genetic engineering in improving crop tolerance to drought. OMICS J. Integr. Biol. 2014, 18, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; Zhang, D.F.; Fu, J.; Shi, Y.S.; Song, Y.C.; Wang, T.Y.; Li, Y. Cloning and characterization of a maize bZIP transcription factor, ZmbZIP72, confers drought and salt tolerance in transgenic Arabidopsis. Planta 2012, 235, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.; Zhang, S.; Yao, J.; Rahman, M.U.; Hanif, M.; Zhu, Y.; Wang, X. Genomic organization of the B3-domain transcription factor family in grapevine (Vitis vinifera L.) and expression during seed development in seedless and seeded cultivars. Int. J. Mol. Sci. 2019, 20, 4553. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, S.M.S.; Deeba, F.; Sultana, T.; Raja, G.K. OsDOF18, A DOF transcription factor from rice confers abiotic stress tolerance in Escherichia coli. Pak. J. Sci. Ind. Res. Ser. Biol. Sci. 2017, 60, 70–79. [Google Scholar] [CrossRef]
- Wang, B.; Li, Z.; Ran, Q.; Li, P.; Peng, Z.; Peng, Z.; Zhang, J. ZmNF-YB16 overexpression improves drought resistance and yield by enhancing photosynthesis and the antioxidant capacity of maize plants. Front. Plant Sci. 2018, 9, 709. [Google Scholar] [CrossRef] [Green Version]
- Danisman, S. TCP transcription factors at the interface between environmental challenges and the plant’s growth responses. Front. Plant Sci. 2016, 7, 1930. [Google Scholar] [CrossRef] [Green Version]
- Kaplan-Levy, R.N.; Brewer, P.B.; Quon, T.; Smyth, D.R. The trihelix family of transcription factors–light, stress and development. Trends Plant Sci. 2012, 17, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.A. Identification of Candidate Resistance Genes in Multiple Herbicide Resistant Echinochloa colona. Ph.D. Dissertation, Mississippi State University, Starkville, MS, USA, January 2017. [Google Scholar]
- Shi, H.; Wang, X.; Ye, T.; Cheng, F.; Deng, J.; Yang, P.; Zhang, Y.; Chan, Z. The Cys2/His2-type zinc finger transcription factor ZAT6 modulates biotic and abiotic stress responses by activating salicylic acid-related genes and CBFs in Arabidopsis. Plant Physiol. 2014, 165, 1367–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Li, J.; Liu, Y.; Li, H. Genome-wide analysis of serine/arginine-rich protein family in wheat and Brachypodium distachyon. Plants 2019, 8, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinabandhu, A. A comprehensive computational analysis of cis-regulatory elements for anthocyanin biosynthesis genes in tomato (Solanum lycopersicum L.). Curr. Biot. 2015, 8, 351–358. [Google Scholar]
- Duan, X.; Yu, Z.; Zang, D.; Zhang, D.; Chen, Y.; Wang, Z.H.; Niu, Y.L.; Wang, Y.C. DNA cis-acting element specifically recognized by ThZFP1 protein. J. Northeast For. Univ. 2018, 46, 35–38. [Google Scholar]
- Guo, C.; Guo, L.; Li, X.; Ma, C.; Duan, W.; Gu, J.; Xu, Z.; Li, R.; Lu, W.; Xiao, K. Transcriptional regulation of the rice phytase gene OsPHY1 by several phytohormones and osmotic stresses using promoter-GUS analysis. Plant Mol. Biol. Rep. 2013, 31, 1461–1473. [Google Scholar] [CrossRef]
- Hatorangan, M.R.; Sentausa, E.; Wijaya, G.Y. In silico identification of cis-regulatory elements of phosphate transporter genes in rice (Oryza sativa L.). J. Crop Sci. Biotechnol. 2009, 12, 25–30. [Google Scholar] [CrossRef]
- Kiran, U.; Abdin, M.Z. Computational predictions of common transcription factor binding sites on the genes of proline metabolism in plants. Bioinformation 2012, 8, 886. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, M.; Sun, J.; Mao, X.; Wang, J.; Liu, H.; Zheng, H.; Li, X.; Zhao, H.; Zou, D. Heavy metal stress-associated proteins in rice and Arabidopsis: Genome-wide identification, phylogenetics, duplication, and expression profiles analysis. Front. Genet. 2020, 11, 477. [Google Scholar] [CrossRef]
- Menezes-Benavente, L.; Teixeira, F.K.; Kamei, C.L.A.; Margis-Pinheiro, M. Salt stress induces altered expression of genes encoding antioxidant enzymes in seedlings of a Brazilian indica rice (Oryza sativa L.). Plant Sci. 2004, 166, 323–331. [Google Scholar] [CrossRef]
- Gotoh, O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 1992, 267, 83–90. [Google Scholar] [CrossRef]
- da Fonseca, R.R.; Antunes, A.; Melo, A.; Ramos, M.J. Structural divergence and adaptive evolution in mammalian cytochromes P450 2C. Gene 2007, 387, 58–66. [Google Scholar] [CrossRef]
- Zawaira, A.; Ching, L.Y.; Coulson, L.; Blackburn, J.; Chun Wei, Y. An expanded, unified substrate recognition site map for mammalian cytochrome P450s: Analysis of molecular interactions between 15 mammalian CYP450 isoforms and 868 substrates. Curr. Drug Metab. 2011, 12, 684–700. [Google Scholar] [CrossRef]
- Wilce, M.; Parker, M.W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta 1994, 1205, 1–18. [Google Scholar] [CrossRef]
- Mohsenzadeh, S.; Esmaeili, M.; Moosavi, F.; Shahrtash, M.; Saffari, B.; Mohabatkar, H. Plant glutathione S-transferase classification, structure and evolution. Afr. J. Biotechnol. 2011, 10, 8160–8165. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Functional Group | NMO | UDPGT | Cytochrome P450 | GST |
|---|---|---|---|---|---|
| Amaranthus hypochondriacus | Dicot | 0 | 0 | 5 | 8 |
| Arabidopsis thaliana | Dicot | 1 | 1 | 0 | 0 |
| Capsicum annuum | Dicot | 1 | 0 | 39 | 24 |
| Citrus clementina | Dicot | 1 | 1 | 48 | 21 |
| Daucus carota | Dicot | 1 | 2 | 26 | 23 |
| Glycine max | Dicot | 1 | 13 | 47 | 39 |
| Ipomoea triloba | Dicot | 1 | 5 | 36 | 20 |
| Prunus persica | Dicot | 1 | 1 | 17 | 31 |
| Vitis vinifera | Dicot | 1 | 2 | 22 | 38 |
| Echinochloa crus-galli | Monocot | 2 | 7 | 0 | 0 |
| Eragrostris curvula | Monocot | 2 | 4 | 23 | 30 |
| Hordeum vulgare | Monocot | 1 | 1 | 71 | 51 |
| Lolium multiflorum | Monocot | 1 | 3 | 0 | 0 |
| Oryza sativa | Monocot | 1 | 3 | 39 | 43 |
| Setaria viridis | Monocot | 2 | 6 | 23 | 22 |
| Sorghum bicolor | Monocot | 2 | 4 | 30 | 28 |
| Triticum aestivum | Monocot | 4 | 18 | 99 | 115 |
| Zea mays | Monocot | 1 | 2 | 22 | 19 |
| Gene | Most Common Motif | |
|---|---|---|
| CYP450 | Motif 1 | GDBFEFIPFGAGRRICPGQNFAL |
| Motif 2 | IKAECKDLFFAGTETTSVTLEWAM | |
| Motif 3 | YLTMIIKETLRLHPPAPLLLP | |
| GST | Motif 1 | TYYFMATPYASLFDAYPHVKAWWEDJMARP |
| Motif 2 | GEHKSPEHLARNPFGQVPALQD | |
| NMO | Motif 1 | CLGTRFVATEESFAHPLYKRKLIEMSCTDYTBVFGRARWPGAPQRVLETP |
| Motif 3 | DHVRELIRKTRSLTEKPFGAAIVLAFPHEENLRVVLEEKLAVLQVYWGEF | |
| Motif 4 | DGIIVQGREAGGHVIGQEGLLPLLPRVVDLVSDSGIPVIAAGGIVDGRGY | |
| Motif 5 | GILGFDYGIVQAPLGPDISGPELAAAVANAGAIGLLRLPDW | |
| UDPGT | Motif 3 | PLHILFFPFLAPGHLIPLADMA |
| Motif 4 | SYGEVFNSFHELEPDYAEHYRT | |
| Motif 7 | RAKELGEKARAAVEEGGSSYNDVGRLIDE | |
| Motif 8 | CTIJTTPVNAAVIRSAVDRAN |
| NTSR Gene | Motif | TFBS |
|---|---|---|
| CYP450 | ABRELATERD1, IDE1 element, SURECOREATSULTR11, IBOXCORE, TAAGSTKST1, ASF1MOTIFCAMV, WBOXHVISO1, BIHD1OS, WBOXATNPR1 CCAATBOX1, DOFCOREZM, WRKY71OS, CACTFTPPCA1, GT1CONSENSUS, GTGANTG10, ARR1AT | GATA; tify, AP2; ERF, Dof, ZF-HD, Homeodomain; TALE, B3, NF-YB, TCP, Trihelix, dehydrin |
| GST | CAATBOX1, CCAATBOX1, IBOXCORE, LTRECOREATCOR15, CGACGOSAMY3, MYBST1, SORLIP1AT, BIHD1OS, GT1CONSENSUS, DOFCOREZM, GTGANTG10, WRKY71OS, CACTFTPPCA1, ARR1AT, EBOXBNNAPA, MYBCOREATCYCB1, ABRELATERD1 | GATA; tify, Dof, AP2; ERF, B3, bHLH, ZF-HD, NF-YB, Trihelix, TCP, Myb/SANT; MYB; ARR-B, SBP, WRKY, bZIP |
| NMO | CAATBOX1, GATABOX, EBOXBNNAPA, GT1CONSENSUS, POLLEN1LELAT52, DOFCOREZM, GTGANTG10, TAAAGSTKST1, MYCCONSENSUSAT, WRKY71OS, CACTFTPPCA1, ARR1AT | GATA; tify, Dof, ZF-HD, Homeodomain; TALE, Myb/SANT; MYB; ARR-B, AP2; ERF, B3, NF-YB, Dehydrin, TCP, Trihelix, bZIP, bHLH, SBP |
| UDPGT | CAATBOX1, GATABOX, DOFCOREZM, WRKY71OS, CACTFTPPCA1, ARR1AT | GATA; tify, Dof, Homeodomain;TALE, AP2; ERF, B3, NF-YB, Dehydrin, TCP, Trihelix, bZIP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandra, S.; Leon, R.G. Genome-Wide Evolutionary Analysis of Putative Non-Specific Herbicide Resistance Genes and Compilation of Core Promoters between Monocots and Dicots. Genes 2022, 13, 1171. https://doi.org/10.3390/genes13071171
Chandra S, Leon RG. Genome-Wide Evolutionary Analysis of Putative Non-Specific Herbicide Resistance Genes and Compilation of Core Promoters between Monocots and Dicots. Genes. 2022; 13(7):1171. https://doi.org/10.3390/genes13071171
Chicago/Turabian StyleChandra, Saket, and Ramon G. Leon. 2022. "Genome-Wide Evolutionary Analysis of Putative Non-Specific Herbicide Resistance Genes and Compilation of Core Promoters between Monocots and Dicots" Genes 13, no. 7: 1171. https://doi.org/10.3390/genes13071171
APA StyleChandra, S., & Leon, R. G. (2022). Genome-Wide Evolutionary Analysis of Putative Non-Specific Herbicide Resistance Genes and Compilation of Core Promoters between Monocots and Dicots. Genes, 13(7), 1171. https://doi.org/10.3390/genes13071171