
Genetic Analysis Algorithm for the Study of Patients with Multiple Congenital Anomalies and Isolated Congenital Heart Disease †

 ,
,  , , , and add
Show full author list
, , , and add
Show full author list
Abstract
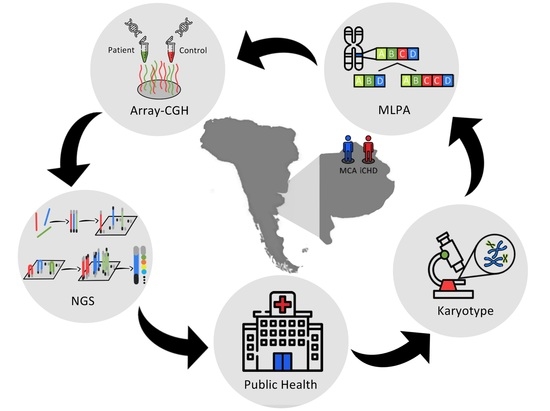
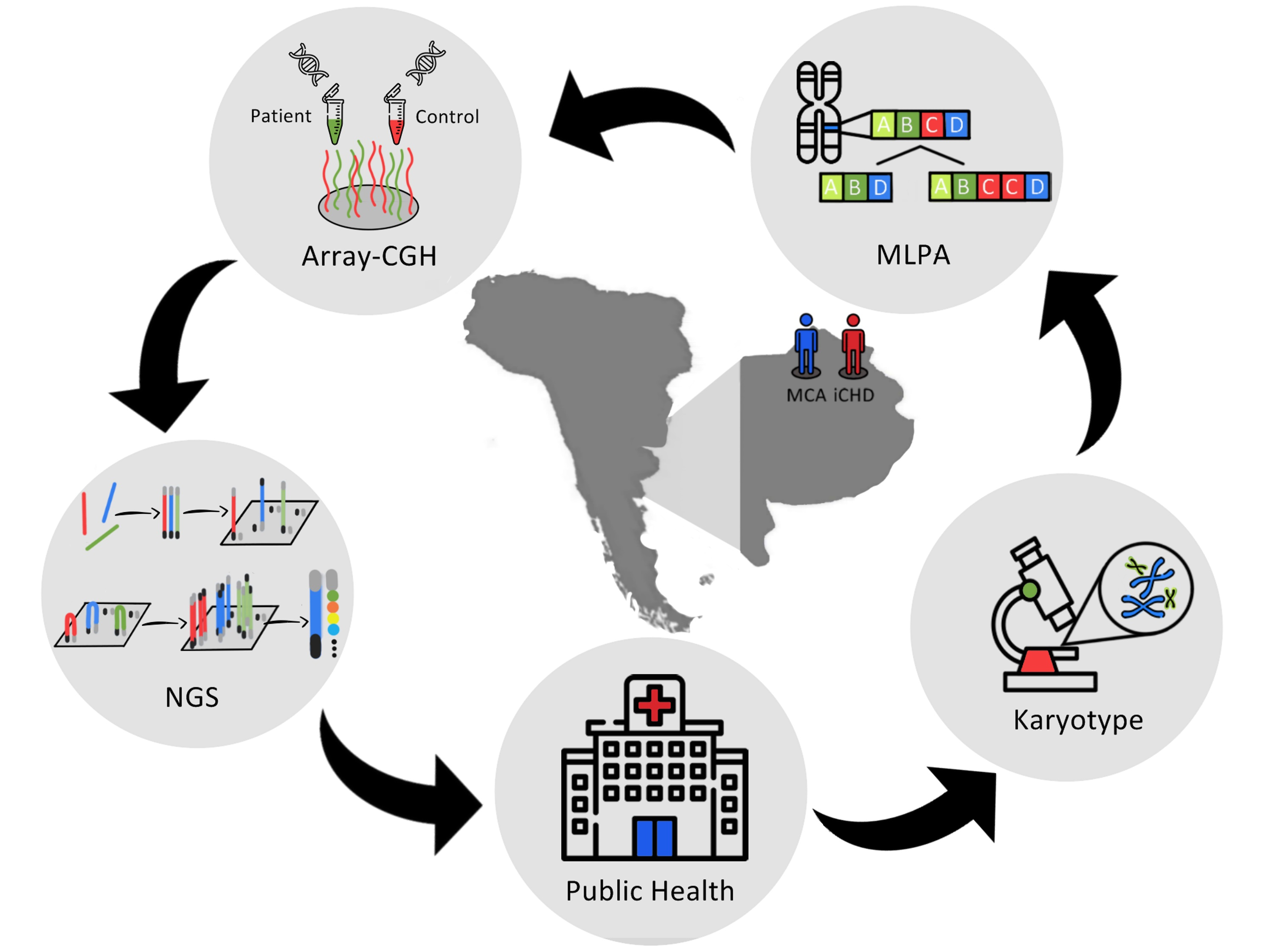
:
1. Introduction
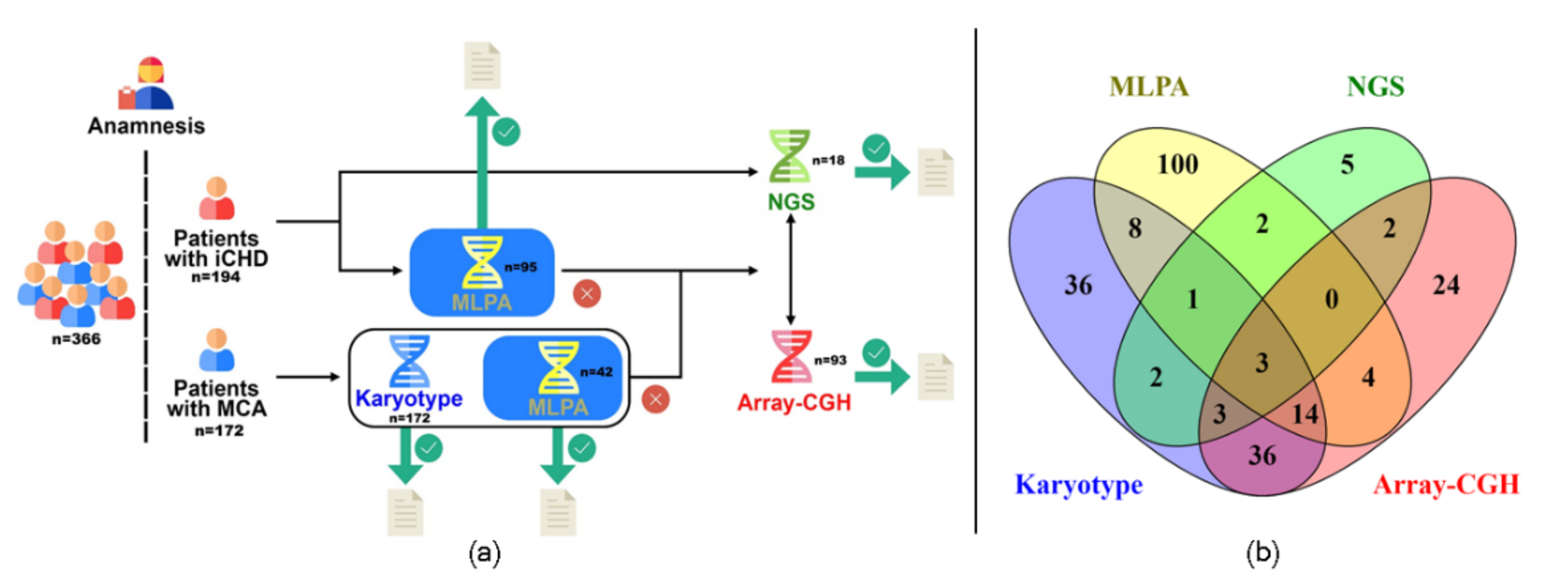
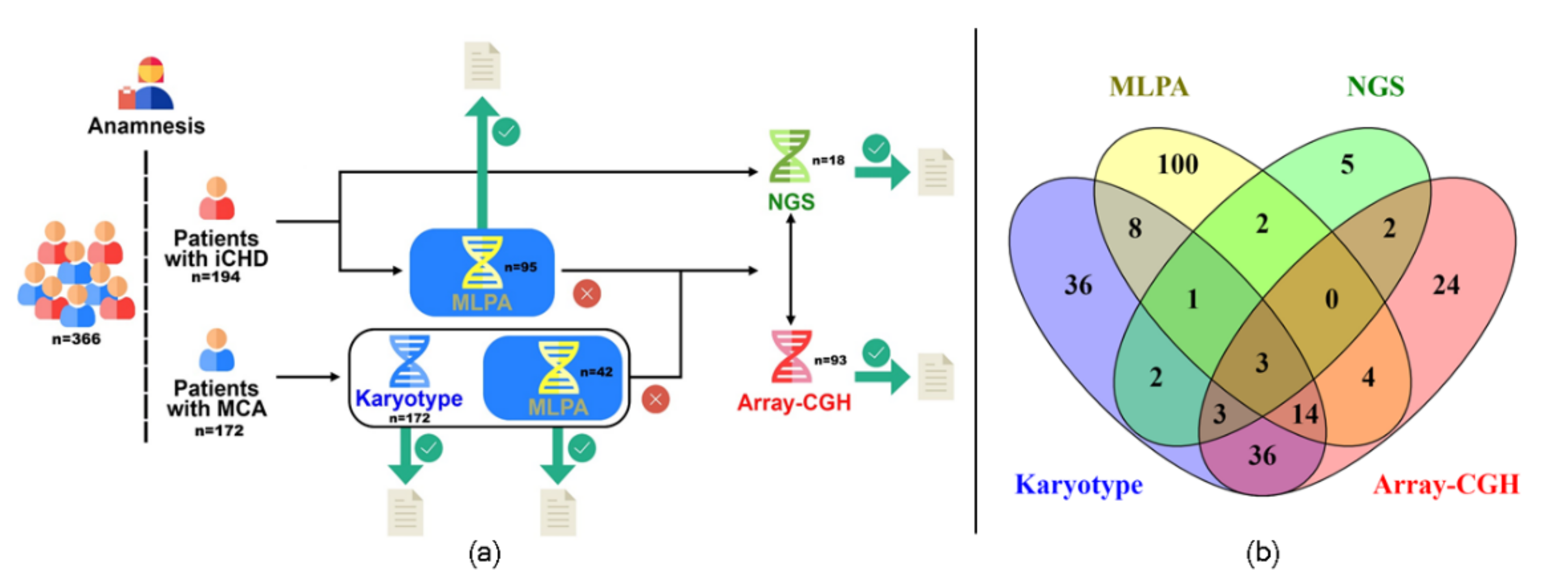
2. Materials and Methods
2.1. Patients
2.2. Algorithm Applied
2.3. Cytogenetic Analysis
2.4. Multiplex Ligation-Dependent Probe Amplification Analysis (MLPA)
2.5. Chromosomal Microarray Analysis (CMA)
2.6. Next-Generation Sequencing (NGS) Analysis
3. Results
3.1. Cytogenetic Analyses
3.2. Multiplex Ligation-Dependent Probe Amplification Analysis (MLPA)
3.3. Chromosomal Microarray Analysis (CMA)
3.4. Next-Generation Sequencing (NGS)

{kind=link}
{kind=link}
| Patient ID | Karyotype | Imbalance | Size (Mb) | Classification | OMIM # | ORPHA # | Supporting Evidence |
|---|---|---|---|---|---|---|---|
| 2 | 46,XX | arr[GRCh37] 2q24.2q31.1(160347642_174075851)x1 | 13.73 | Pathogenic | - | 1617 | [37,38] |
| 41 | 46,XY | arr[GRCh37] 7q11.23(72766313_74042787)x3 | 1.27 | Pathogenic | 609757 | 261102 | [39] |
| 48 | 46,XY,t(1;2)(q25;q21) 1 | arr[GRCh37] 8q21.11q21.3(75904944_87097083)x1 | 11.19 | Pathogenic | 614230 | 284160 | [40] |
| 65 | Failed | arr[GRCh37] 7q36.1q36.3(149062717_159124131)x1 | 10.06 | Pathogenic | - | [41,42,43] | |
| 68 | Failed | arr[GRCh37] 2q14.2q14.3(120628484_127658188)x1 | 7 | Pathogenic | [44] | ||
| 94 | 47,XXX [28]/47,XX,+14 [12] | arr(14)x3,(X)x3 | - | Pathogenic | - | - | [31] |
| 96 | 46,XX | arr[GRCh37] Xp22.33(940688_2676609)x3 | 1.7 | Pathogenic | - | - | [45] |
| 100 2 | Failed | arr[GRCh37] 16p13.11(15551302_16194578)x1pat | 0.64 | Pathogenic | 619351 | 2241 | [46,47] |
| 106 | 46,XX | arr[GRCh37] 17q25.3(80583397_81044553)x1 | 0.46 | Likely Pathogenic | - | [48,49] | |
| 127 | 46,XY | arr[GRCh37] 16p12.2(21837492_22407931)x1 | 0.57 | Pathogenic | 136570 | [50] | |
| 134 | Failed | arr[GRCh37] 1p36.33p36.23(834101_7930605)x1; 7q35q36.3(146927174_159128556)x3 1,3 | 7.1; 12.2 | Pathogenic | 607872,- | 1606 | [51,52,53,54] |
| 147 | 46,XY | arr[GRCh37] 15q14(33809650_40027263)x1 | 6.22 | Pathogenic | 616898 | 261190 | [55,56] |
| 149 4 | 46, XY | arr[GRCh37] 5q22.2(112155123_112174165)x1pat | 0.02 | Pathogenic | - | 261584 | [57,58] |
| 167 | Failed | arr(13)x3 | - | Pathogenic | - | 3378 | [59] |
| 187 | Failed | arr(18)x3 | - | Pathogenic | - | 3380 | [60] |
| 233 | 46,XY | arr[GRCh37] 9q22.2q31.1(93864974_106661581)x1 | 12 | Pathogenic | 109400 | [61] | |
| 362 | Failed | arr[GRCh37] 3p21.31(44948482_49115809)x1dn | 4.1 | Pathogenic | - | - | [62,63,64,65] |
| 368 | 46,XY,trp(8)(p21.1p21.2) | arr[GRCh37] 8p21.3p21.2(19779604_26531980)x4 | 6.7 | Pathogenic | - | - | [66] |
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention (CDC). Update on overall prevalence of major birth defects–Atlanta: Georgia, 1978–2005. Morb. Mortal. Wkly. Rep. 2008, 57, 1–5. [Google Scholar]
- Rasmussen, S.A.; Olney, R.S.; Holmes, L.B.; Lin, A.E.; Keppler-Noreuil, K.M.; Moore, C.A. National birth defects prevention study guidelines for case classification for the national birth defects prevention study. Birth Defects Res. A Clin. Mol. Teratol. 2003, 67, 193–201. [Google Scholar] [CrossRef]
- Pan American Health Organization. Present and Future of Birth Defects Surveillance in the Americas; Pan American Health Organization: Washington, DC, USA, 2019; ISBN 9789275121924. [Google Scholar]
- Van Der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [Green Version]
- Baird, P.A.; Anderson, T.W.; Newcombe, H.B.; Lowry, R.B. Genetic disorders in children and young adults: A population study. Am. J. Hum. Genet. 1988, 42, 677–693. [Google Scholar]
- Szczałuba, K.; Nowakowska, B.; Sobecka, K.; Smyk, M.; Castaneda, J.; Klapecki, J.; Kutkowska-Kaźmierczak, A.; Śmigiel, R.; Bocian, E.; Radkowski, M.; et al. Application of array comparative genomic hybridization in newborns with multiple congenital anomalies. Adv. Exp. Med. Biol. 2016, 912, 1–9. [Google Scholar] [CrossRef]
- Szczałuba, K.; Demkow, U. Array comparative genomic hybridization and genomic sequencing in the diagnostics of the causes of congenital anomalies. J. Appl. Genet. 2017, 58, 185–198. [Google Scholar] [CrossRef]
- Groisman, B.; Bidondo, M.P.; Barbero, P.; Gili, J.A.; Liascovich, R. RENAC: Registro Nacional de Anomalías Congénitas de Argentina. Archivos Argentinos De Pediatría 2013, 111, 484–494. [Google Scholar]
- Agopian, A.J.; Evans, J.A.; Lupo, P.J. Analytic methods for evaluating patterns of multiple congenital anomalies in birth defect registries. Birth Defects Res. 2018, 110, 5–11. [Google Scholar] [CrossRef]
- Crotwell, P.L.; Hoyme, H.E. Advances in whole-genome genetic testing: From chromosomes to microarrays. Curr. Probl. Pediatr. Adolesc. Health Care 2012, 42, 47–73. [Google Scholar] [CrossRef]
- Michelson, D.J.; Clark, R.D. Optimizing genetic diagnosis of neurodevelopmental disorders in the clinical setting. Clin. Lab. Med. 2020, 40, 231–256. [Google Scholar] [CrossRef]
- Wellesley, D.; Dolk, H.; Boyd, P.A.; Greenlees, R.; Haeusler, M.; Nelen, V.; Garne, E.; Khoshnood, B.; Doray, B.; Rissmann, A.; et al. Rare chromosome abnormalities, prevalence and prenatal diagnosis rates from population-based congenital anomaly registers in Europe. Eur. J. Hum. Genet. 2012, 20, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.-Y.; Phung, M.T.; Shaw, C.A.; Pham, K.; Neil, S.E.; Patel, A.; Sahoo, T.; Bacino, C.A.; Stankiewicz, P.; Kang, S.-H.L.; et al. Genomic imbalances in neonates with birth defects: High detection rates by using chromosomal microarray analysis. Pediatrics 2008, 122, 1310–1318. [Google Scholar] [CrossRef] [Green Version]
- Valduga, M.; Philippe, C.; Bach Segura, P.; Thiebaugeorges, O.; Miton, A.; Beri, M.; Bonnet, C.; Nemos, C.; Foliguet, B.; Jonveaux, P. A retrospective study by oligonucleotide array-CGH analysis in 50 fetuses with multiple malformations. Prenat. Diagn. Publ. Affil. Int. Soc. Prenat. Diagn. 2010, 30, 333–341. [Google Scholar] [CrossRef]
- Ziolkowska, L.; Kawalec, W.; Turska-Kmiec, A.; Krajewska-Walasek, M.; Brzezinska-Rajszys, G.; Daszkowska, J.; Maruszewski, B.; Burczynski, P. Chromosome 22q11.2 microdeletion in children with conotruncal heart defects: Frequency, associated cardiovascular anomalies, and outcome following Cardiac surgery. Eur. J. Pediatr. 2008, 167, 1135–1140. [Google Scholar] [CrossRef]
- Poirsier, C.; Besseau-Ayasse, J.; Schluth-Bolard, C.; Toutain, J.; Missirian, C.; Le Caignec, C.; Bazin, A.; De Blois, M.C.; Kuentz, P.; Catty, M.; et al. A French multicenter study of over 700 patients with 22q11 deletions diagnosed using FISH or aCGH. Eur. J. Hum. Genet. 2016, 24, 844–851. [Google Scholar] [CrossRef] [Green Version]
- Mlynarski, E.E.; Xie, M.; Taylor, D.; Sheridan, M.B.; Guo, T.; Racedo, S.E.; McDonald-McGinn, D.M.; Chow, E.W.C.; Vorstman, J.; Swillen, A.; et al. Rare copy number variants and congenital heart defects in the 22q11.2 deletion syndrome. Hum. Genet. 2016, 135, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, R.E.; Hall, J.G.; Everman, D.B.; Solomon, B.D. (Eds.) Human Malformations and Related Anomalies. In Oxford Monographs on Medical Genetics, 3rd ed.; Oxford University Press: New York, NY, USA, 2015; ISBN 9780199386031. [Google Scholar]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic basis for congenital heart disease: Revisited: A scientific statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef]
- Shashi, V.; McConkie-Rosell, A.; Rosell, B.; Schoch, K.; Vellore, K.; McDonald, M.; Jiang, Y.-H.; Xie, P.; Need, A.; Goldstein, D.B. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet. Med. 2014, 16, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Boycott, K.M.; Hartley, T.; Biesecker, L.G.; Gibbs, R.A.; Innes, A.M.; Riess, O.; Belmont, J.; Dunwoodie, S.L.; Jojic, N.; Lassmann, T.; et al. A Diagnosis for all rare genetic diseases: The horizon and the next frontiers. Cell 2019, 177, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Krepischi-Santos, A.C.V.; Vianna-Morgante, A.M.; Jehee, F.S.; Passos-Bueno, M.R.; Knijnenburg, J.; Szuhai, K.; Sloos, W.; Mazzeu, J.F.; Kok, F.; Cheroki, C.; et al. Whole-genome array-CGH screening in undiagnosed syndromic patients: Old syndromes revisited and new alterations. Cytogenet. Genome Res. 2006, 115, 254–261. [Google Scholar] [CrossRef]
- Campos, C.M.R.; Zanardo, E.A.; Dutra, R.L.; Kulikowski, L.D.; Kim, C.A. Investigation of copy number variation in children with conotruncal heart defects. Arq. Bras. Cardiol. 2015, 104, 24–31. [Google Scholar] [CrossRef]
- De Souza, K.R.; Mergener, R.; Huber, J.; Campos Pellanda, L.; Riegel, M. Cytogenomic evaluation of subjects with syndromic and nonsyndromic conotruncal heart defects. Biomed. Res. Int. 2015, 2015, 401941. [Google Scholar] [CrossRef]
- Lay-Son, G.; Espinoza, K.; Vial, C.; Rivera, J.C.; Guzmán, M.L.; Repetto, G.M. Chromosomal microarrays testing in children with developmental disabilities and congenital anomalies. J. Pediatr. 2015, 91, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Vianna, G.S.; Medeiros, P.F.V.; Alves, A.F.; Silva, T.O.; Jehee, F.S. Array-CGH analysis in patients with intellectual disability and/or congenital malformations in Brazil. Genet. Mol. Res. 2016, 15, 15017769. [Google Scholar] [CrossRef]
- Zanardo, É.A.; Dutra, R.L.; Piazzon, F.B.; Dias, A.T.; Novo-Filho, G.M.; Nascimento, A.M.; Montenegro, M.M.; Damasceno, J.G.; Madia, F.A.R.; da Costa, T.V.M.M.; et al. Cytogenomic assessment of the diagnosis of 93 patients with developmental delay and multiple congenital abnormalities: The Brazilian experience. Clinics 2017, 72, 526–537. [Google Scholar] [CrossRef]
- Delea, M.; Espeche, L.D.; Bruque, C.D.; Bidondo, M.P.; Massara, L.S.; Oliveri, J.; Brun, P.; Cosentino, V.R.; Martinoli, C.; Tolaba, N.; et al. Genetic imbalances in Argentinean patients with congenital conotruncal heart defects. Genes 2018, 9, 454. [Google Scholar] [CrossRef] [Green Version]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. (Eds.) International Standing Committee on Human Cytogenomic Nomenclature. In ISCN 2020: An International System for Human Cytogenomic Nomenclature (2020); Karger: Basel, Switzerland, 2020; ISBN 9783318067064. [Google Scholar]
- Espeche, L.D.; Solari, A.P.; Mori, M.Á.; Arenas, R.M.; Palomares, M.; Pérez, M.; Martínez, C.; Lotersztein, V.; Segovia, M.; Armando, R.; et al. Implementation of chromosomal microarrays in a cohort of patients with intellectual disability at the Argentinean public health system. Mol. Biol. Rep. 2020, 47, 6863–6878. [Google Scholar] [CrossRef]
- Massara, L.S.; Delea, M.; Espeche, L.; Bruque, C.D.; Oliveri, J.; Brun, P.; Furforo, L.; Dain, L.; Rozental, S. Double autosomal/gonosomal mosaic trisomy 47,XXX/47,XX,+14 in a newborn with multiple congenital anomalies. Cytogenet. Genome Res. 2019, 159, 137–142. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American college of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Pua, C.J.; Bhalshankar, J.; Miao, K.; Walsh, R.; John, S.; Lim, S.Q.; Chow, K.; Buchan, R.; Soh, B.Y.; Lio, P.M.; et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J. Cardiovasc. Transl. Res. 2016, 9, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the Association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; Mcgowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. Hgvs recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [Green Version]
- McElhinney, D.B.; McDonald-McGinn, D.; Zackai, E.H.; Goldmuntz, E. Cardiovascular anomalies in patients diagnosed with a chromosome 22q11 deletion beyond 6 months of age. Pediatrics 2001, 108, e104. [Google Scholar] [CrossRef] [Green Version]
- Davidsson, J.; Collin, A.; Olsson, M.E.; Lundgren, J. Deletion of the SCN gene cluster on 2q24. 4 is associated with severe epilepsy: An array-based genotype–phenotype correlation and a comprehensive review of previously published cases. Epilepsy Res. 2008, 81, 69–79. [Google Scholar] [CrossRef]
- Krepischi, A.C.V.; Knijnenburg, J.; Bertola, D.R.; Kim, C.A.; Pearson, P.L.; Bijlsma, E.; Szuhai, K.; Kok, F.; Vianna-Morgante, A.M.; Rosenberg, C. Two distinct regions in 2q24.2-q24.3 associated with idiopathic epilepsy: Epilepsy associated with 2q24 deletions. Epilepsia 2010, 51, 2457–2460. [Google Scholar] [CrossRef] [Green Version]
- Mervis, C.B.; Morris, C.A.; Klein-Tasman, B.P.; Velleman, S.L.; Osborne, L.R. 7q11.23 Duplication Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2015. [Google Scholar]
- Palomares, M.; Delicado, A.; Mansilla, E.; de Torres, M.L.; Vallespín, E.; Fernandez, L.; Martinez-Glez, V.; García-Miñaur, S.; Nevado, J.; Simarro, F.S.; et al. Characterization of a 8q21.11 microdeletion syndrome associated with intellectual disability and a recognizable phenotype. Am. J. Hum. Genet. 2011, 89, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Lukusa, T.; Vermeesch, J.R.; Fryns, J.P. De novo deletion 7q36 resulting from a distal 7q/8q translocation: Phenotypic expression and comparison to the literature. Genet. Couns. 2005, 16, 1–15. [Google Scholar]
- Roessler, E.; El-Jaick, K.B.; Dubourg, C.; Vélez, J.I.; Solomon, B.D.; Pineda-Álvarez, D.E.; Lacbawan, F.; Zhou, N.; Ouspenskaia, M.; Paulussen, A.; et al. The mutational spectrum of holoprosencephaly-associated changes within the SHH gene in humans predicts loss-of-function through either key structural alterations of the ligand or its altered synthesis. Human Mutat. 2009, 30, E921–E935. [Google Scholar] [CrossRef] [Green Version]
- Ayub, S.; Gadji, M.; Krabchi, K.; Côté, S.; Gekas, J.; Maranda, B.; Drouin, R. Three new cases of terminal deletion of the long arm of chromosome 7 and literature review to correlate genotype and phenotype manifestations. Am. J. Med. Genet. A 2016, 170A, 896–907. [Google Scholar] [CrossRef]
- Kordaß, U.; Schröder, C.; Elbracht, M.; Soellner, L.; Eggermann, T. A familial GLI2 deletion (2q14.2) Not associated with the holoprosencephaly syndrome phenotype. Am. J. Med. Genet. Part A 2015, 167, 1121–1124. [Google Scholar] [CrossRef]
- Bunyan, D.J.; Baffico, M.; Capone, L.; Vannelli, S.; Iughetti, L.; Schmitt, S.; Taylor, E.-J.; Herridge, A.A.; Shears, D.; Forabosco, A.; et al. Duplications upstream and downstream of SHOX identified as novel causes of leri-weill dyschondrosteosis or idiopathic short stature. Am. J. Med. Genet. Part A 2016, 170, 949–957. [Google Scholar] [CrossRef]
- Kloth, K.; Renner, S.; Burmester, G.; Steinemann, D.; Pabst, B.; Lorenz, B.; Simon, R.; Kolbe, V.; Hempel, M.; Rosenberger, G. 16p13.11 Microdeletion Uncovers loss-of-function of a MYH11 missense variant in a patient with megacystis-microcolon-intestinal-hypoperistalsis syndrome. Clin. Genet. 2019, 96, 85–90. [Google Scholar] [CrossRef]
- Ambartsumyan, L. Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome Overview. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Probst, F.J.; James, R.A.; Burrage, L.C.; Rosenfeld, J.A.; Bohan, T.P.; Ward Melver, C.H.; Magoulas, P.; Austin, E.; Franklin, A.I.A.; Azamian, M.; et al. De novo deletions and duplications of 17q25.3 cause susceptibility to cardiovascular malformations. Orphanet J. Rare Dis. 2015, 10, 75. [Google Scholar] [CrossRef] [Green Version]
- Miyake, N.; Fukai, R.; Ohba, C.; Chihara, T.; Miura, M.; Shimizu, H.; Kakita, A.; Imagawa, E.; Shiina, M.; Ogata, K.; et al. Biallelic TBCD mutations cause early-onset neurodegenerative encephalopathy. Am. J. Hum. Genet. 2016, 99, 950–961. [Google Scholar] [CrossRef] [Green Version]
- Girirajan, S.; Pizzo, L.; Moeschler, J.; Rosenfeld, J. 16p12.2 Recurrent deletion. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2015; 16p. [Google Scholar]
- Forabosco, A.; Baroncini, A.; Dalpra, L.; Chessa, L.; Giannotti, A.; Maccagnani, F.; Dallapiccola, B. The phenotype of partial dup(7q) reconsidered: A report of five new cases. Clin. Genet. 1988, 34, 48–59. [Google Scholar] [CrossRef]
- Morava, E.; Bartsch, O.; Czakó, M.; Frensel, A.; Kalscheuer, V.; Kárteszi, J.; Kosztolányi, G. Small inherited terminal duplication of 7q with hydrocephalus, cleft palate, joint contractures, and severe hypotonia. Clin. Dysmorphol. 2003, 12, 123–127. [Google Scholar] [CrossRef]
- Jordan, V.K.; Zaveri, H.P.; Scott, D.A. 1p36 Deletion syndrome: An update. Appl. Clin. Genet. 2015, 8, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Fernández, M.-C.; Ramírez-Oyaga, S.; Restrepo, C.M.; Huertas-Quiñones, V.-M.; Barrera-Castañeda, M.; Quero, R.; Hernández-Toro, C.-J.; Tamar Silva, C.; Laissue, P.; Cabrera, R. Identification of a New Candidate locus for ebstein anomaly in 1p36.2. Mol. Syndromol. 2018, 9, 164–169. [Google Scholar] [CrossRef]
- Erdogan, F.; Ullmann, R.; Chen, W.; Schubert, M.; Adolph, S.; Hultschig, C.; Kalscheuer, V.; Ropers, H.-H.; Spaich, C.; Tzschach, A. Characterization of a 5.3 Mb Deletion in 15q14 by comparative genomic hybridization using a whole genome ″Tiling Path″ BAC array in a girl with heart defect, cleft palate, and developmental delay. Am. J. Med. Genet. A 2007, 143A, 172–178. [Google Scholar] [CrossRef]
- Nevado, J.; Mergener, R.; Palomares-Bralo, M.; Souza, K.R.; Vallespín, E.; Mena, R.; Martínez-Glez, V.; Mori, M.Á.; Santos, F.; García-Miñaur, S.; et al. New microdeletion and microduplication syndromes: A comprehensive review. Genet. Mol. Biol. 2014, 37, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Quadri, M.; Vetro, A.; Gismondi, V.; Marabelli, M.; Bertario, L.; Sala, P.; Varesco, L.; Zuffardi, O.; Ranzani, G.N. APC Rearrangements in familial adenomatous polyposis: Heterogeneity of deletion lengths and breakpoint sequences underlies similar phenotypes. Fam. Cancer 2015, 14, 41–49. [Google Scholar] [CrossRef]
- Michils, G.; Tejpar, S.; Thoelen, R.; Van Cutsem, E.; Vermeesch, J.R.; Fryns, J.-P.; Legius, E.; Matthijs, G. Large deletions of the APC gene in 15% of mutation-negative patients with classical polyposis (FAP): A Belgian study. Hum. Mutat. 2005, 25, 125–134. [Google Scholar] [CrossRef]
- Williams, G.M.; Brady, R. Patau Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Cereda, A.; Carey, J.C. The Trisomy 18 Syndrome. Orphanet J. Rare Dis. 2012, 7, 81. [Google Scholar] [CrossRef] [Green Version]
- Muller, E.A.; Aradhya, S.; Atkin, J.F.; Carmany, E.P.; Elliott, A.M.; Chudley, A.E.; Clark, R.D.; Everman, D.B.; Garner, S.; Hall, B.D.; et al. Microdeletion 9q22.3 syndrome includes metopic craniosynostosis, hydrocephalus, macrosomia, and developmental delay. Am. J. Med. Genet. Part A 2012, 158A, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Eto, K.; Sakai, N.; Shimada, S.; Shioda, M.; Ishigaki, K.; Hamada, Y.; Shinpo, M.; Azuma, J.; Tominaga, K.; Shimojima, K.; et al. Microdeletions of 3p21.31 characterized by developmental delay, distinctive features, elevated serum creatine kinase levels, and white matter involvement. Am. J. Med. Genet. A 2013, 161, 3049–3056. [Google Scholar] [CrossRef]
- Haldeman-Englert, C.R.; Gai, X.; Perin, J.C.; Ciano, M.; Halbach, S.S.; Geiger, E.A.; McDonald-McGinn, D.M.; Hakonarson, H.; Zackai, E.H.; Shaikh, T.H. A 3.1-Mb Microdeletion of 3p21.31 associated with cortical blindness, cleft lip, CNS abnormalities, and developmental delay. Eur. J. Med. Genet. 2009, 52, 265–268. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Hwang, H.; Kim, S.Y.; Kim, K.J.; Choi, J.S.; Woo, M.J.; Choi, Y.M.; Jun, J.K.; Lim, B.C.; Chae, J.H. Chromosomal microarray with clinical diagnostic utility in children with developmental delay or intellectual disability. Ann. Lab. Med. 2018, 38, 473–480. [Google Scholar] [CrossRef]
- Lovrecic, L.; Bertok, S.; Žerjav Tanšek, M. A New case of an extremely rare 3p21.31 interstitial deletion. Mol. Syndromol. 2016, 7, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.T.; Harikumar, C.; Fisher, R.B. Agenesis of the corpus callosum in mosaic tetrasomy 8p. Clin. Dysmorphol. 2010, 19, 215–217. [Google Scholar] [CrossRef]
- Lee, J.J.; Ekker, S.C.; Von Kessler, D.P.; Porter, J.A.; Sun, B.I.; Beachy, P.A. Autoproteolysis in hedgehog protein biogenesis. Science 1994, 266, 1528–1537. [Google Scholar] [CrossRef]
- Yetman, A.T.; Starr, L.J. Newly Described Recessive MYH11 Disorder with clinical overlap of multisystemic smooth muscle dysfunction and megacystis microcolon hypoperistalsis syndromes. Am. J. Med. Genet. A 2018, 176, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Nagai, T.; Hasegawa, T.; Kinoshita, E.; Tanaka, T.; Ogata, T. Two NOVEL AND ONE recurrentPTPN11 Mutations in LEOPARD Syndrome. Am. J. Med. Genet. 2004, 130, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Kontaridis, M.I.; Swanson, K.D.; David, F.S.; Barford, D.; Neel, B.G. PTPN11 (Shp2) Mutations in LEOPARD syndrome have dominant negative, not activating, effects. J. Biol. Chem. 2006, 281, 6785–6792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Babu, M.; Raghunath, A.; Venkatesh, C.P. Genetic analysis of a five generation indian family with BPES: A novel missense mutation (p.Y215C). Mol. Vis. 2004, 10, 445–449. [Google Scholar] [PubMed]
- Nallathambi, J.; Laissue, P.; Batista, F.; Benayoun, B.A.; Lesaffre, C.; Moumné, L.; Pandaranayaka, P.E.; Usha, K.; Krishnaswamy, S.; Sundaresan, P.; et al. Differential functional effects of novel mutations of the transcription factor FOXL2 in BPES patients. Hum. Mutat. 2008, 29, E123–E131. [Google Scholar] [CrossRef]
- Kitsiou-Tzeli, S.; Papadopoulou, A.; Kanaka-Gantenbein, C.; Fretzayas, A.; Daskalopoulos, D.; Kanavakis, E.; Nicolaidou, P. Does the rare A172G mutation of PTPN11 gene convey a mild noonan syndrome phenotype? Horm. Res. 2006, 66, 124–131. [Google Scholar] [CrossRef]
- Fergelot, P.; Van Belzen, M.; Van Gils, J.; Afenjar, A.; Armour, C.M.; Arveiler, B.; Beets, L.; Burglen, L.; Busa, T.; Collet, M.; et al. Phenotype and genotype in 52 patients with rubinstein-taybi syndrome caused by EP300 mutations. Am. J. Med. Genet. A 2016, 170, 3069–3082. [Google Scholar] [CrossRef]
- Menke, L.A.; Gardeitchik, T.; Hammond, P.; Heimdal, K.R.; Houge, G.; Hufnagel, S.B.; Ji, J.; Johansson, S.; Kant, S.G.; Kinning, E.; et al. Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling rubinstein-taybi syndrome. Am. J. Med. Genet. A 2018, 176, 862–876. [Google Scholar] [CrossRef]
- Stevens, C.A. Rubinstein-Taybi Syndrome Synonym: Broad Thumb-Hallux Syndrome. Gene 2004, 10. [Google Scholar] [CrossRef]
- Tartaglia, M.; Kalidas, K.; Shaw, A.; Song, X.; Musat, D.L.; van der Burgt, I.; Brunner, H.G.; Bertola, D.R.; Crosby, A.; Ion, A.; et al. PTPN11 Mutations in noonan syndrome: Molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 2002, 70, 1555–1563. [Google Scholar] [CrossRef] [Green Version]
- Strullu, M.; Caye, A.; Lachenaud, J.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Pouvreau, N.; Pereira, S.; Baumann, C.; Contet, A.; et al. Juvenile myelomonocytic leukaemia and noonan syndrome. J. Med. Genet. 2014, 51, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Joyce, S.; Gordon, K.; Brice, G.; Ostergaard, P.; Nagaraja, R.; Short, J.; Moore, S.; Mortimer, P.; Mansour, S. The lymphatic phenotype in noonan and cardiofaciocutaneous syndrome. Eur. J. Hum. Genet. 2016, 24, 690–696. [Google Scholar] [CrossRef] [Green Version]
- Van Trier, D.C.; Vos, A.M.C.; Draaijer, R.W.; Van Der Burgt, I.; Draaisma, J.M.T.; Cruysberg, J.R.M. Ocular manifestations of noonan syndrome: A prospective clinical and genetic study of 25 patients. Ophthalmology 2016, 123, 2137–2146. [Google Scholar] [CrossRef]
- Mendez, R.; Delea, M.; Dain, L.; Rittler, M. A novel pathogenic frameshift variant of KAT6B identified by clinical exome sequencing in a newborn with the say–barber–biesecker–young–simpson syndrome. Clin. Dysmorphol. 2020, 29, 42. [Google Scholar] [CrossRef] [PubMed]
- LaHaye, S.; Corsmeier, D.; Basu, M.; Bowman, J.L.; Fitzgerald-Butt, S.; Zender, G.; Bosse, K.; McBride, K.L.; White, P.; Garg, V. Utilization of whole exome sequencing to identify causative mutations in familial congenital heart disease. Circ. Cardiovasc. Genet. 2016, 9, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunert, M.; Dorn, C.; Schueler, M.; Dunkel, I.; Schlesinger, J.; Mebus, S.; Alexi-Meskishvili, V.; Perrot, A.; Wassilew, K.; Timmermann, B.; et al. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated tetralogy of fallot. Hum. Mol. Genet. 2014, 23, 3115–3128. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, C.; Astrea, G.; Savarese, M.; Cassandrini, D.; Brisca, G.; Trucco, F.; Pedemonte, M.; Trovato, R.; Ruggiero, L.; Vercelli, L.; et al. MYH7-Related myopathies: Clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J. Rare Dis. 2016, 11, 91. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Aoki, Y.; Niihori, T.; Cavé, H.; Verloes, A.; Okamoto, N.; Kawame, H.; Fujiwara, I.; Takada, F.; Ohata, T.; et al. Molecular and clinical analysis of RAF1 in noonan syndrome and related disorders: Dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum. Mutat. 2010, 31, 284–294. [Google Scholar] [CrossRef]
- Komissarova, S.M.; Rineiska, N.M.; Chakova, N.N.; Niyazova, S.S. Overlapping phenotype: Left ventricular non-compaction and hypertrophic cardiomyopathy. Kardiologiia 2020, 60, 137–145. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Nucaro, A.L.; Rossino, R.; Pruna, D.; Rassu, S.; Cianchetti, C.; Cao, A.; Moi, P. Prenatal diagnosis of a Mosaic supernumerary marker iso (8p) (tetrasomy 8p): Discordance between chorionic villi culture and amniotic fluid karyotypes. Prenat. Diagn. 2006, 26, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.V.P.F.; Gamba, B.F.; Empke, S.L.L.; Alves, C.C.D.O.; Bérgamo, N.A.; Ribeiro-Bicudo, L.A. Congenital heart disease revealing familial 22q11 deletion syndrome. Int. J. Cardiovasc. Sci. 2020, 33, 425–426. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071. [Google Scholar] [CrossRef] [Green Version]
- Szczałuba, K.; Nowakowska, B.A.; Sobecka, K.; Smyk, M.; Castaneda, J.; Dudkiewicz, Z.; Kutkowska-Kaźmierczak, A.; Sąsiadek, M.M.; Śmigiel, R.; Bocian, E. High-resolution array comparative genomic hybridization utility in Polish newborns with isolated cleft lip and palate. Neonatology 2015, 107, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.E.; Geiger, E.; James, A.C.; Ciprero, K.L.; Nimmakayalu, M.; Zhang, Y.; Huang, A.; Vaddi, M.; Rappaport, E.; Zackai, E.H.; et al. Rapid detection of submicroscopic chromosomal rearrangements in children with multiple congenital anomalies using high density oligonucleotide arrays. Hum. Mutat. 2006, 27, 467–473. [Google Scholar] [CrossRef]
- Correia-Costa, G.R.; Sgardioli, I.C.; Santos, A.P.D.; Araujo, T.K.D.; Secolin, R.; Lopes-Cendes, I.; Gil-da-Silva-Lopes, V.L.; Vieira, T.P. Increased runs of homozygosity in the autosomal genome of Brazilian Individuals with neurodevelopmental delay/intellectual disability and/or multiple congenital Anomalies Investigated by chromosomal microarray analysis. Genet. Mol. Biol. 2022, 45, e20200480. [Google Scholar] [CrossRef]
- Jang, W.; Kim, Y.; Han, E.; Park, J.; Chae, H.; Kwon, A.; Choi, H.; Kim, J.; Son, J.O.; Lee, S.J.; et al. Chromosomal microarray analysis as a first-tier clinical diagnostic test in patients with developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies: A prospective multicenter study in Korea. Ann. Lab. Med. 2019, 39, 299–310. [Google Scholar] [CrossRef]
- Çebi, A.H.; Altıner, Ş. Application of chromosome microarray analysis in the investigation of developmental disabilities and congenital anomalies: Single center experience and review of and deletions. Mol. Syndromol. 2020, 11, 197–206. [Google Scholar] [CrossRef]
- Cheng, S.S.W.; Chan, K.Y.K.; Leung, K.K.P.; Au, P.K.C.; Tam, W.-K.; Li, S.K.M.; Luk, H.-M.; Kan, A.S.Y.; Chung, B.H.Y.; Lo, I.F.M.; et al. Experience of chromosomal microarray applied in prenatal and postnatal settings in Hong Kong. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 196–207. [Google Scholar] [CrossRef]
- Yokoi, T.; Enomoto, Y.; Tsurusaki, Y.; Harada, N.; Saito, T.; Nagai, J.-I.; Naruto, T.; Kurosawa, K. An efficient genetic test flow for multiple congenital anomalies and intellectual disability. Pediatr. Int. 2020, 62, 556–561. [Google Scholar] [CrossRef]
- Bertola, D.R.; Castro, M.A.A.; Yamamoto, G.L.; Honjo, R.S.; Ceroni, J.R.; Buscarilli, M.M.; Freitas, A.B.; Malaquias, A.C.; Pereira, A.C.; Jorge, A.A.L.; et al. Phenotype-genotype analysis of 242 individuals with RASopathies: 18-year experience of a tertiary center in Brazil. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 31851. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, L.A.; Pires, L.V.L.; Yamamoto, G.L.; Ceroni, M., Jr.; Honjo, R.S.; De Novaes França Bisneto, E.; Oliveira, L.A.N.; Rosenberg, C.; Krepischi, A.C.V.; Passos-Bueno, M.R.; et al. Congenital limb deficiency: Genetic investigation of 44 individuals presenting mainly longitudinal defects in isolated or syndromic forms. Clin. Genet. 2021, 100, 14041. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Bernardini, L.; Lepri, F.; Giuffrida, M.G.; Guida, V.; Baban, A.; Versacci, P.; Capolino, R.; Torres, B.; De Luca, A.; et al. Ebstein anomaly: Genetic heterogeneity and association with microdeletions 1p36 and 8p23.1. Am. J. Med. Genet. A 2011, 155, 2196–2202. [Google Scholar] [CrossRef] [PubMed]
- Furey, C.G.; Choi, J.; Jin, S.C.; Zeng, X.; Timberlake, A.T.; Nelson-Williams, C.; Mansuri, M.S.; Lu, Q.; Duran, D.; Panchagnula, S.; et al. De novo mutation in genes regulating neural stem cell fate in human congenital hydrocephalus. Neuron 2018, 99, 302–314.e4. [Google Scholar] [CrossRef] [Green Version]
- Guterman, S.; Beneteau, C.; Redon, S.; Dupont, C.; Missirian, C.; Jaeger, P.; Herve, B.; Jacquin, C.; Douet-Guilbert, N.; Till, M.; et al. Prenatal findings in 1p36 deletion syndrome: New cases and a literature review. Prenat. Diagn. 2019, 39, 871–882. [Google Scholar] [CrossRef]
- Aristidou, C.; Theodosiou, A.; Bak, M.; Mehrjouy, M.M.; Constantinou, E.; Alexandrou, A.; Papaevripidou, I.; Christophidou-Anastasiadou, V.; Skordis, N.; Kitsiou-Tzeli, S.; et al. Position effect, cryptic complexity, and direct gene disruption as disease mechanisms in de novo apparently balanced translocation cases. PLoS ONE 2018, 13, e0205298. [Google Scholar] [CrossRef] [Green Version]
- Matsumaru, D.; Haraguchi, R.; Miyagawa, S.; Motoyama, J.; Nakagata, N.; Meijlink, F.; Yamada, G. Genetic analysis of hedgehog signaling in ventral body wall development and the onset of omphalocele formation. PLoS ONE 2011, 6, e16260. [Google Scholar] [CrossRef] [Green Version]
- Tabin, C.J.; McMahon, A.P. Recent advances in hedgehog signalling. Trends Cell Biol. 1997, 7, 442–446. [Google Scholar] [CrossRef]
- Ming, J.E.; Roessler, E.; Muenke, M. Human Developmental Disorders and the Sonic Hedgehog Pathway. Mol. Med. Today 1998, 4, 343–349. [Google Scholar] [CrossRef]
- Kim, P.C.; Mo, R.; Hui, C.C. Murine Models of VACTERL Syndrome: Role of sonic hedgehog signaling pathway. J. Pediatr. Surg. 2001, 36, 381–384. [Google Scholar] [CrossRef]
- Monzani, A.; Babu, D.; Mellone, S.; Genoni, G.; Fanelli, A.; Prodam, F.; Bellone, S.; Giordano, M. Co-occurrence of genomic imbalances on Xp22.1 in the SHOX region and 15q25.2 in a girl with short stature, precocious puberty, urogenital malformations and bone anomalies. BMC Med. Genom. 2019, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glessner, J.T.; Bick, A.G.; Ito, K.; Homsy, J.G.; Rodriguez-Murillo, L.; Fromer, M.; Mazaika, E.; Vardarajan, B.; Italia, M.; Leipzig, J.; et al. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ. Res. 2014, 115, 884–896. [Google Scholar] [CrossRef]
- Sandoval, J.I.; De Jesus, O. Hydranencephaly; StatPearls: Treasure Island, FL, USA, 2022. Available online: https://pubmed.ncbi.nlm.nih.gov/32644417/ (accessed on 20 June 2022).
- Lewis, S.A.; Tian, G.; Cowan, N.J. The α- and β-tubulin folding pathways. Trends Cell Biol. 1997, 7, 479–484. [Google Scholar] [CrossRef]
- Yokoi, S.; Ishihara, N.; Miya, F.; Tsutsumi, M.; Yanagihara, I.; Fujita, N.; Yamamoto, H.; Kato, M.; Okamoto, N.; Tsunoda, T.; et al. TUBA1A Mutation can cause a hydranencephaly-like severe form of cortical dysgenesis. Sci. Rep. 2015, 5, 5–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirozzi, F.; Nelson, B.; Mirzaa, G. From microcephaly to megalencephaly: Determinants of brain size. Dialogues Clin. Neurosci. 2018, 20, 267–282. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical Mechanisms of Vertebrate Hedgehog Signaling. Development 2019, 146, 166892. [Google Scholar] [CrossRef] [Green Version]
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, J.; Wang, H.; Feng, Q.; Luo, F.; Xie, J. Compound heterozygous variants in MYH11 underlie autosomal recessive megacystis-microcolon-intestinal hypoperistalsis syndrome in a Chinese family. J. Hum. Genet. 2019, 64, 1067–1073. [Google Scholar] [CrossRef]
- Zhu, L.; Vranckx, R.; Van Kien, P.K.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.-M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic Aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef]
- Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Scherer, S.; Liu, Y.; Presley, C.; Guo, D.; Estrera, A.L.; Safi, H.J.; Brasier, A.R.; et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007, 16, 2453–2462. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, J.; Ouled Amar Bencheikh, B.; Hamdan, F.F.; Harrison, S.M.; Baker, L.A.; Couture, F.; Thiffault, I.; Ouazzani, R.; Samuels, M.E.; Mitchell, G.A.; et al. A homozygous loss-of-function variant in MYH11 in a case with megacystis-microcolon-intestinal hypoperistalsis syndrome. Eur. J. Hum. Genet. 2015, 23, 1266–1268. [Google Scholar] [CrossRef] [Green Version]
- López, M.; Seidel, V.; Santibáñez, P.; Cervera-Acedo, C.; Castro-de Castro, P.; Domínguez-Garrido, E. First case report of inherited rubinstein-taybi syndrome associated with a novel EP300 variant. BMC Med. Genet. 2016, 17, 97. [Google Scholar] [CrossRef] [Green Version]
- García-Castro, M.; Coto, E.; Reguero, J.R.; Berrazueta, J.R.; Álvarez, V.; Alonso, B.; Sainz, R.; Martín, M.; Morís, C. Mutations in sarcomeric genes MYH7, MYBPC3, TNNT2, TNNI3, and TPM1 in patients with hypertrophic cardiomyopathy. Rev. Española Cardiol. 2009, 62, 48–56. [Google Scholar] [CrossRef]
- Coto, E.; Palacín, M.; Martín, M.; Castro, M.G.; Reguero, J.R.; García, C.; Berrazueta, J.R.; Morís, C.; Morales, B.; Ortega, F.; et al. Functional polymorphisms in genes of the angiotensin and serotonin systems and Risk of hypertrophic cardiomyopathy: AT1R as a potential modifier. J. Transl. Med. 2010, 8, 1–9. [Google Scholar] [CrossRef]
- Sanchez, O.; Campuzano, O.; Fernández-Falgueras, A.; Sarquella-Brugada, G.; Cesar, S.; Mademont, I.; Mates, J.; Pérez-Serra, A.; Coll, M.; Pico, F.; et al. Natural and undetermined sudden death: Value of post-mortem genetic investigation. PLoS ONE 2016, 11, 167358. [Google Scholar] [CrossRef]
- Gómez, J.; Lorca, R.; Reguero, J.R.; Morís, C.; Martín, M.; Tranche, S.; Alonso, B.; Iglesias, S.; Alvarez, V.; Díaz-Molina, B.; et al. Screening of the Filamin C gene in a large cohort of hypertrophic cardiomyopathy patients. Circ. Cardiovasc. Genet. 2017, 10, e001584. [Google Scholar] [CrossRef] [Green Version]
- Sedaghat-Hamedani, F.; Kayvanpour, E.; Tugrul, O.F.; Lai, A.; Amr, A.; Haas, J.; Proctor, T.; Ehlermann, P.; Jensen, K.; Katus, H.A.; et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: A meta-analysis on 7675 individuals. Clin. Res. Cardiol. 2018, 107, 30–41. [Google Scholar] [CrossRef]
- Wang, L.; Geist, J.; Grogan, A.; Hu, L.R.; Kontrogianni-Konstantopoulos, A. Thick Filament Protein Network, Functions, and Disease Association. Compr. Physiol. 2018, 8, 170023. [Google Scholar] [CrossRef]
| Karyotype | N | N (%) |
|---|---|---|
| 46,XX | 51 | 89 (86.4) |
| 46, XY | 38 | |
| 47,XY,+13 | 1 | 13 (12.6) |
| 47,XY,+18 or 47,XX,+18 | 6 | |
| 47,XXX [28]/47,XX,+14 [12] 1,2 | 1 | |
| 46,XY,t(1;2)(q25;q21) 1 | 1 | |
| 46,XX,t(11;17)(p10;p10) | 1 | |
| 46,XX,del(15)(q11.2q13) 3 | 1 | |
| 47,XY,+idic(18)(p10) 3 | 1 | |
| 46,XY,trp(8)(p21.2p21.1) 1 | 1 | |
| Total | 103 | 103 (100) |
| Imbalances | Conotruncal CHD | Suspected 22q11DS 1 | Total | ||
|---|---|---|---|---|---|
| With MCA | Isolated CHD | With MCA | Isolated CHD | ||
| None | 18 | 60 | 15 | 12 | 105 |
| del(22)(q11) 3 Mb | 5 | 13 | - | 3 | 21 |
| del(22)(q11) 1.5 Mb | 1 | 2 | - | - | 3 |
| dup(22)(q11) 1.5 Mb | 1 | 1 | - | - | 2 |
| rsa22q11.2 (TBX1-7x1) | 1 | - | - | 1 | |
| Total | 25 | 77 | 15 | 15 | 132 |
| Patient ID | Phenotype | Gene | Sequencing Technology | Genetic Variant | Protein Change | Classification | Supporting Evidence |
|---|---|---|---|---|---|---|---|
| 57 | MCA | SHH | WES | (NM_000193.4):c.808C>T | p.His270Tyr | Likely Pathogenic | [67] |
| 100 | MCA | MYH11 | WES | (NM_001040114.1):c.3143-2_3145delAGTGC | p.? | Pathogenic | [47,68] |
| 114 | MCA | PTPN11 | TSC | (NM_002834.5):c.1381G>A | p.(Ala461Thr) | Pathogenic | [69,70] |
| 123 | MCA | FOXL2 | WES | (NM_023067.4):c.644A>G | p.(Tyr215Cys) | Pathogenic | [71,72] |
| 129 | MCA | PTPN11 | WES | (NM_002834.5):c.922A>G | p.Asn308Asp | Pathogenic | [73] |
| 149 | MCA | EP300 | WES | (NM_001429.4):c.7081C>T | p.(Gln2361Ter) | Pathogenic | [74,75,76] |
| 175 | MCA | PTPN11 | TSC | (NM_002834.35)c.181G>A | p.(Asp61Asn) | Pathogenic | [77,78,79,80] |
| 188 | MCA | KAT6B | WES | (NM_012330.4):c.4572_4573dupTA | p.(Thr1525IlefsTer25) | Pathogenic | [81] |
| 232 1 | MCA | MYBPC3 | WES | (NM_000256.3):c.2176C>T | p.(Arg726Cys) | Likely Pathogenic | [82,83] |
| 333 | iCHD | MYH7 | TSC | (NM_000257.4):c.671A>T | p.(Asn224Ile) | Likely Pathogenic | [84] |
| 335 | iCHD | RAF1 | TSC | (NM_002880.3):c.770C>T | p.(Ser257Leu) | Pathogenic | [85] |
| 351 1 | iCHD | MYBPC3 | TSC | (NM_000256.3):c.2176C>T | p.(Arg726Cys) | Likely Pathogenic | [86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delea, M.; Massara, L.S.; Espeche, L.D.; Bidondo, M.P.; Barbero, P.; Oliveri, J.; Brun, P.; Fabro, M.; Galain, M.; Fernández, C.S.; et al. Genetic Analysis Algorithm for the Study of Patients with Multiple Congenital Anomalies and Isolated Congenital Heart Disease. Genes 2022, 13, 1172. https://doi.org/10.3390/genes13071172
Delea M, Massara LS, Espeche LD, Bidondo MP, Barbero P, Oliveri J, Brun P, Fabro M, Galain M, Fernández CS, et al. Genetic Analysis Algorithm for the Study of Patients with Multiple Congenital Anomalies and Isolated Congenital Heart Disease. Genes. 2022; 13(7):1172. https://doi.org/10.3390/genes13071172
Chicago/Turabian StyleDelea, Marisol, Lucia S. Massara, Lucia D. Espeche, María Paz Bidondo, Pablo Barbero, Jaen Oliveri, Paloma Brun, Mónica Fabro, Micaela Galain, Cecilia S. Fernández, and et al. 2022. "Genetic Analysis Algorithm for the Study of Patients with Multiple Congenital Anomalies and Isolated Congenital Heart Disease" Genes 13, no. 7: 1172. https://doi.org/10.3390/genes13071172
APA StyleDelea, M., Massara, L. S., Espeche, L. D., Bidondo, M. P., Barbero, P., Oliveri, J., Brun, P., Fabro, M., Galain, M., Fernández, C. S., Taboas, M., Bruque, C. D., Kolomenski, J. E., Izquierdo, A., Berenstein, A., Cosentino, V., Martinoli, C., Vilas, M., Rittler, M., ... on behalf of the PID ACM-CC Group. (2022). Genetic Analysis Algorithm for the Study of Patients with Multiple Congenital Anomalies and Isolated Congenital Heart Disease. Genes, 13(7), 1172. https://doi.org/10.3390/genes13071172