
A Novel MTTK Gene Variant m.8315A>C as a Cause of MERRF Syndrome

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient
2.2. Mitochondrial DNA Sequencing
2.3. Mitochondria Isolation
2.4. MEGS Analysis
2.5. Spectrophotometry
2.6. Electrophoresis
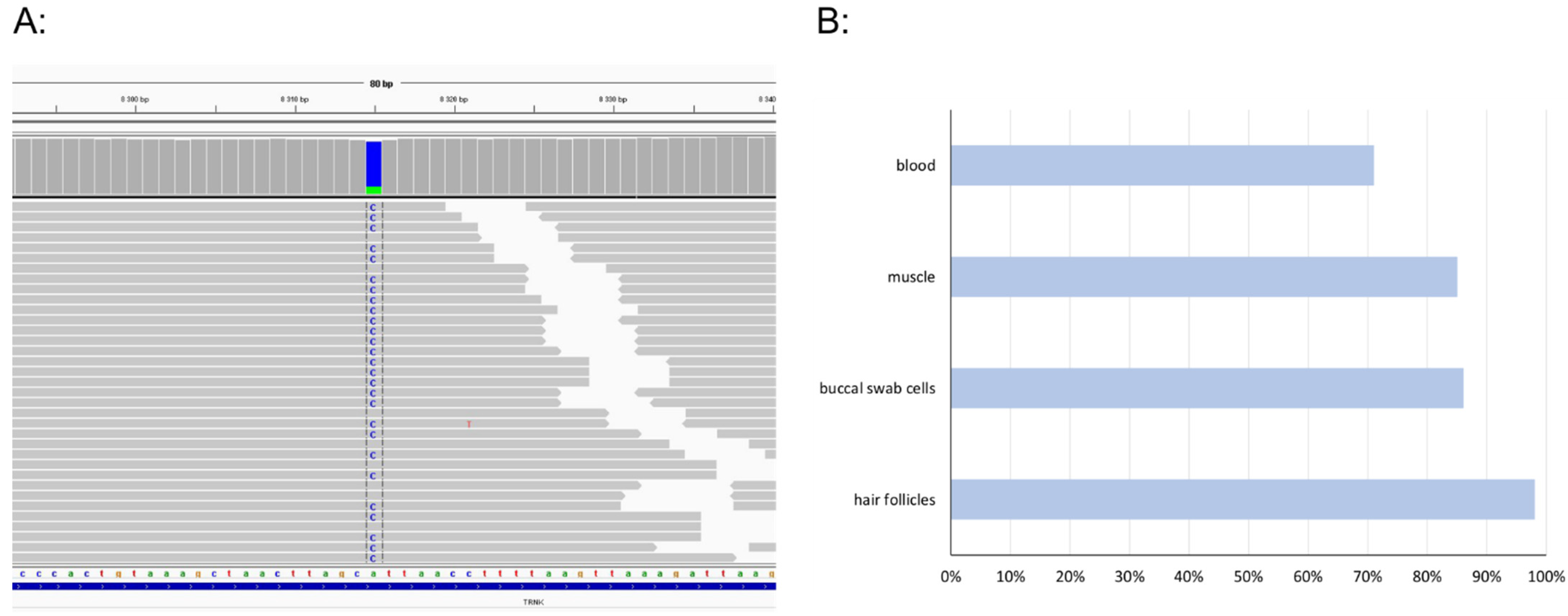
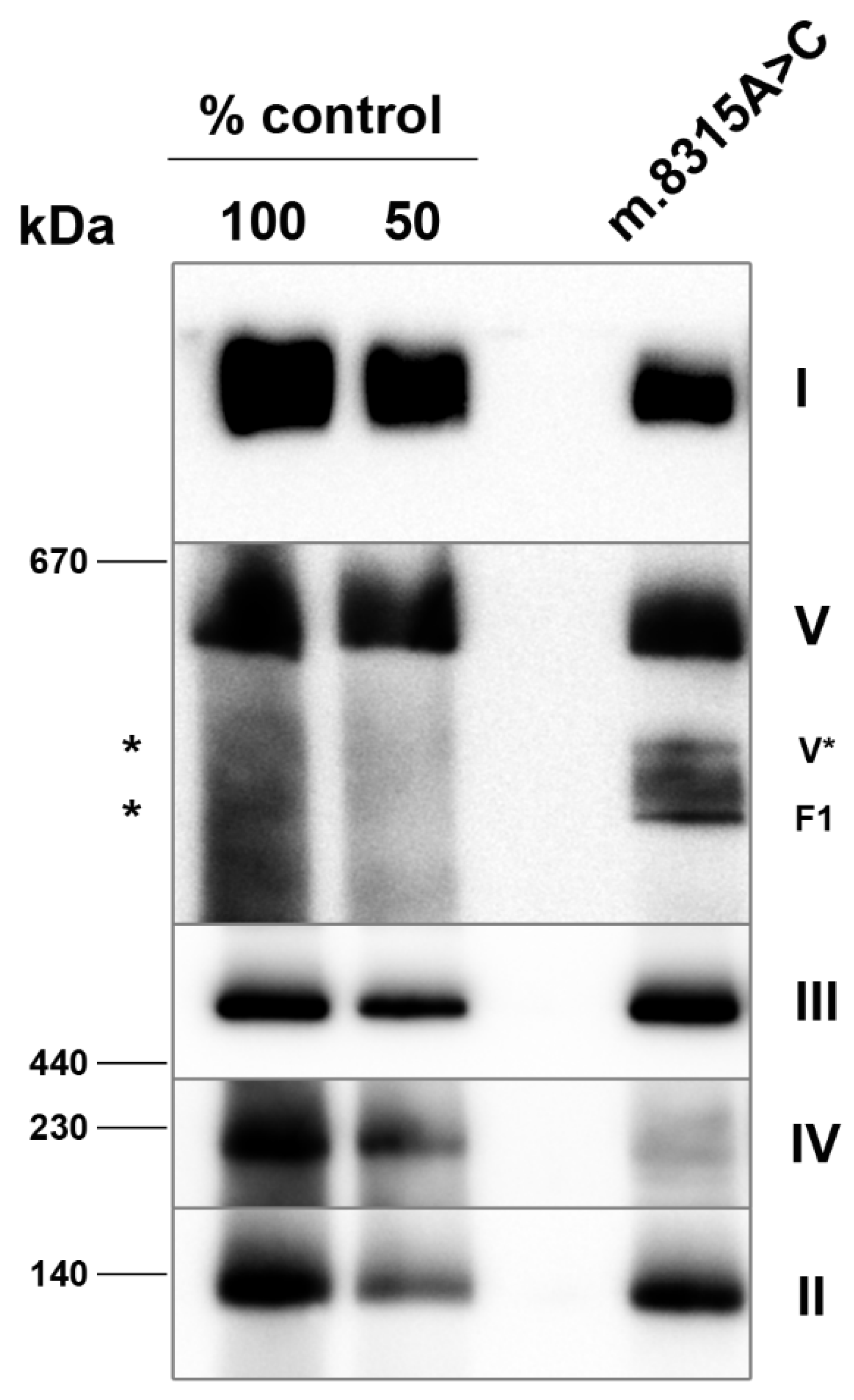
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Richter, U.; McFarland, R.; Taylor, R.W.; Pickett, S.J. The molecular pathology of pathogenic mitochondrial tRNA variants. FEBS Lett. 2021, 595, 1003–1024. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MITOMAP: A Human Mitochondrial Genome Database 2019. Available online: http://www.mitomap.org (accessed on 26 May 2022).
- Blakely, E.L.; Alston, C.L.; Lecky, B.; Chakrabarti, B.; Falkous, G.; Turnbull, D.M.; Taylor, R.W.; Gorman, G.S. Distal weakness with respiratory insufficiency caused by the m.8344A>G “MERRF” mutation. Neuromuscul. Disord. 2014, 24, 533–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Minetti, C.; Moggio, M.; Mongini, T.; Servidei, S.; et al. Phenotypic heterogeneity of the 8344A>G mtDNA “MERRF” mutation. Neurology 2013, 80, 2049–2054. [Google Scholar] [CrossRef] [PubMed]
- Makinen, M.W.; Lee, C.P.; Shy, G.M. Microanalysis of cytochrome content, oxidative and phosphorylative activities of human skeletal muscle mitochondria. Neurology 1968, 18, 299. [Google Scholar] [PubMed]
- Jesina, P.; Tesarova, M.; Fornuskova, D.; Vojtiskova, A.; Pecina, P.; Kaplanova, V.; Hansikova, H.; Zeman, J.; Houstek, J. Diminished synthesis of subunit a (ATP6) and altered function of ATP synthase and cytochrome c oxidase due to the mtDNA 2 bp microdeletion of TA at positions 9205 and 9206. Biochem. J. 2004, 383, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, A.J.; Trijbels, F.J.; Sengers, R.C.; Wintjes, L.T.; Ruitenbeek, W.; Smeitink, J.A.; Morava, E.; van Engelen, B.G.; van den Heuvel, L.P.; Rodenburg, R.J. Measurement of the energy-generating capacity of human muscle mitochondria: Diagnostic procedure and application to human pathology. Clin. Chem. 2006, 52, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Rustin, P.; Chretien, D.; Bourgeron, T.; Gerard, B.; Rotig, A.; Saudubray, J.M.; Munnich, A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta 1994, 228, 35–51. [Google Scholar] [CrossRef]
- Srere, P.A.; John, M.L. Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)]. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1969; pp. 3–11. [Google Scholar]
- Schagger, H.; Cramer, W.A.; von Jagow, G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 1994, 217, 220–230. [Google Scholar] [CrossRef]
- McCormick, E.M.; Lott, M.T.; Dulik, M.C.; Shen, L.; Attimonelli, M.; Vitale, O.; Karaa, A.; Bai, R.; Pineda-Alvarez, D.E.; Singh, L.N.; et al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum. Mutat. 2020, 41, 2028–2057. [Google Scholar] [CrossRef] [PubMed]
- Blakely, E.L.; Yarham, J.W.; Alston, C.L.; Craig, K.; Poulton, J.; Brierley, C.; Park, S.M.; Dean, A.; Xuereb, J.H.; Anderson, K.N.; et al. Pathogenic mitochondrial tRNA point mutations: Nine novel mutations affirm their importance as a cause of mitochondrial disease. Hum. Mutat. 2013, 34, 1260–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putz, J.; Dupuis, B.; Sissler, M.; Florentz, C. Mamit-tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA 2007, 13, 1184–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laricchia, K.M.; Lake, N.J.; Watts, N.A.; Shand, M.; Haessly, A.; Gauthier, L.; Benjamin, D.; Banks, E.; Soto, J.; Garimella, K.; et al. Mitochondrial DNA variation across 56,434 individuals in gnomAD. Genome Res. 2022, 32, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Bolze, A.; Mendez, F.; White, S.; Tanudjaja, F.; Isaksson, M.; Jiang, R.; Rossi, A.D.; Cirulli, E.; Rashkin, M.; Metcalf, W.; et al. A catalog of homoplasmic and heteroplasmic mitochondrial DNA variants in humans. bioRxiv 2019. [Google Scholar] [CrossRef]
- Mancuso, M.; Petrozzi, L.; Filosto, M.; Nesti, C.; Rocchi, A.; Choub, A.; Pistolesi, S.; Massetani, R.; Fontanini, G.; Siciliano, G. MERRF syndrome without ragged-red fibers: The need for molecular diagnosis. Biochem. Biophys. Res. Commun. 2007, 354, 1058–1060. [Google Scholar] [CrossRef] [PubMed]
- Fornuskova, D.; Brantova, O.; Tesarova, M.; Stiburek, L.; Honzik, T.; Wenchich, L.; Tietzeova, E.; Hansikova, H.; Zeman, J. The impact of mitochondrial tRNA mutations on the amount of ATP synthase differs in the brain compared to other tissues. Biochim. Biophys. Acta 2008, 1782, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Patient | Controls (n = 26) | |
|---|---|---|
| (nmol/min/mg Protein) | ||
| Complex I | 262.1 | 118–282 |
| Complex I + III | 50.7 | 100–287 |
| Complex II | 124.8 | 28–94 |
| Complex II + III | 320 | 97–267 |
| Complex III | 620.8 | 321–640 |
| Complex IV | 112.9 | 825–1500 |
| Citrate Synthase (CS) | 573.3 | 528–938 |
| Complex IV/CS | 0.20 | 1.16–2.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štufková, H.; Kolářová, H.; Lokvencová, K.; Honzík, T.; Zeman, J.; Hansíková, H.; Tesařová, M. A Novel MTTK Gene Variant m.8315A>C as a Cause of MERRF Syndrome. Genes 2022, 13, 1245. https://doi.org/10.3390/genes13071245
Štufková H, Kolářová H, Lokvencová K, Honzík T, Zeman J, Hansíková H, Tesařová M. A Novel MTTK Gene Variant m.8315A>C as a Cause of MERRF Syndrome. Genes. 2022; 13(7):1245. https://doi.org/10.3390/genes13071245
Chicago/Turabian StyleŠtufková, Hana, Hana Kolářová, Kateřina Lokvencová, Tomáš Honzík, Jiří Zeman, Hana Hansíková, and Markéta Tesařová. 2022. "A Novel MTTK Gene Variant m.8315A>C as a Cause of MERRF Syndrome" Genes 13, no. 7: 1245. https://doi.org/10.3390/genes13071245