
The Complete Mitochondrial Genome of Ophioglossum vulgatum L. Is with Highly Repetitive Sequences: Intergenomic Fragment Transfer and Phylogenetic Analysis


Abstract
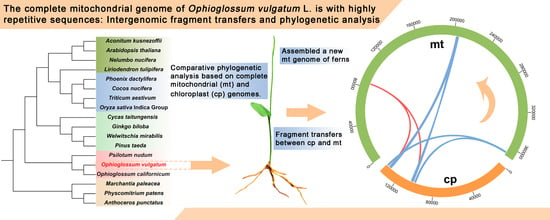
:
1. Introduction
2. Materials and Methods
2.1. DNA Extraction, Illumina DNA Library Construction and Sequencing
2.2. Assembly and Annotation of the mt Genome
2.3. Analysis of mt Genome Characters
2.4. Phylogenetic and Fragment Transfer Analysis of mt and cp Genomes
2.5. Identification and Analysis of Repetitive Sequences
3. Results
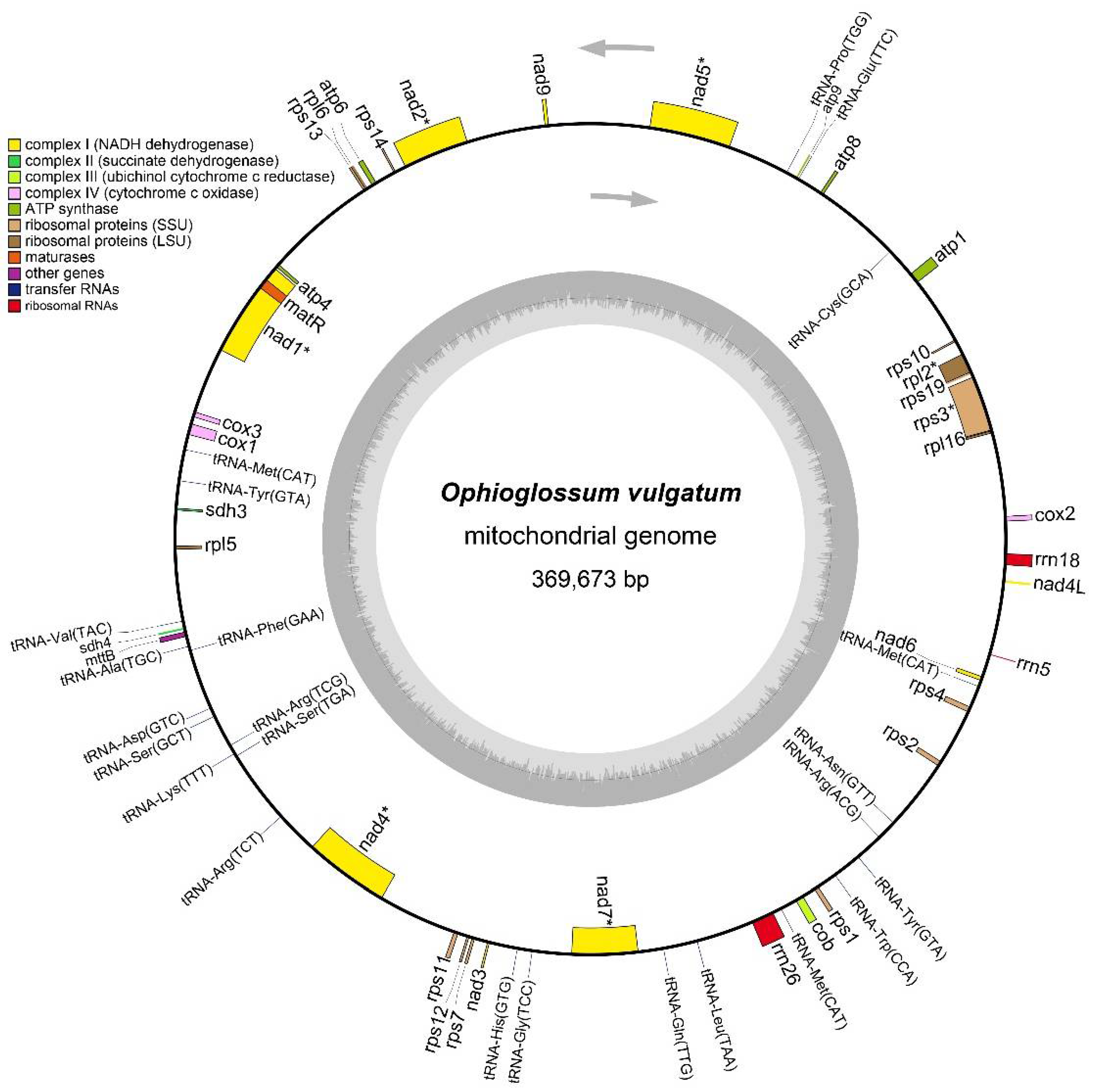
3.1. Characters of the Complete mt Genome of O. vulgatum
3.2. Comparative mt Genomic Analysis between O. vulgatum and O. californicum
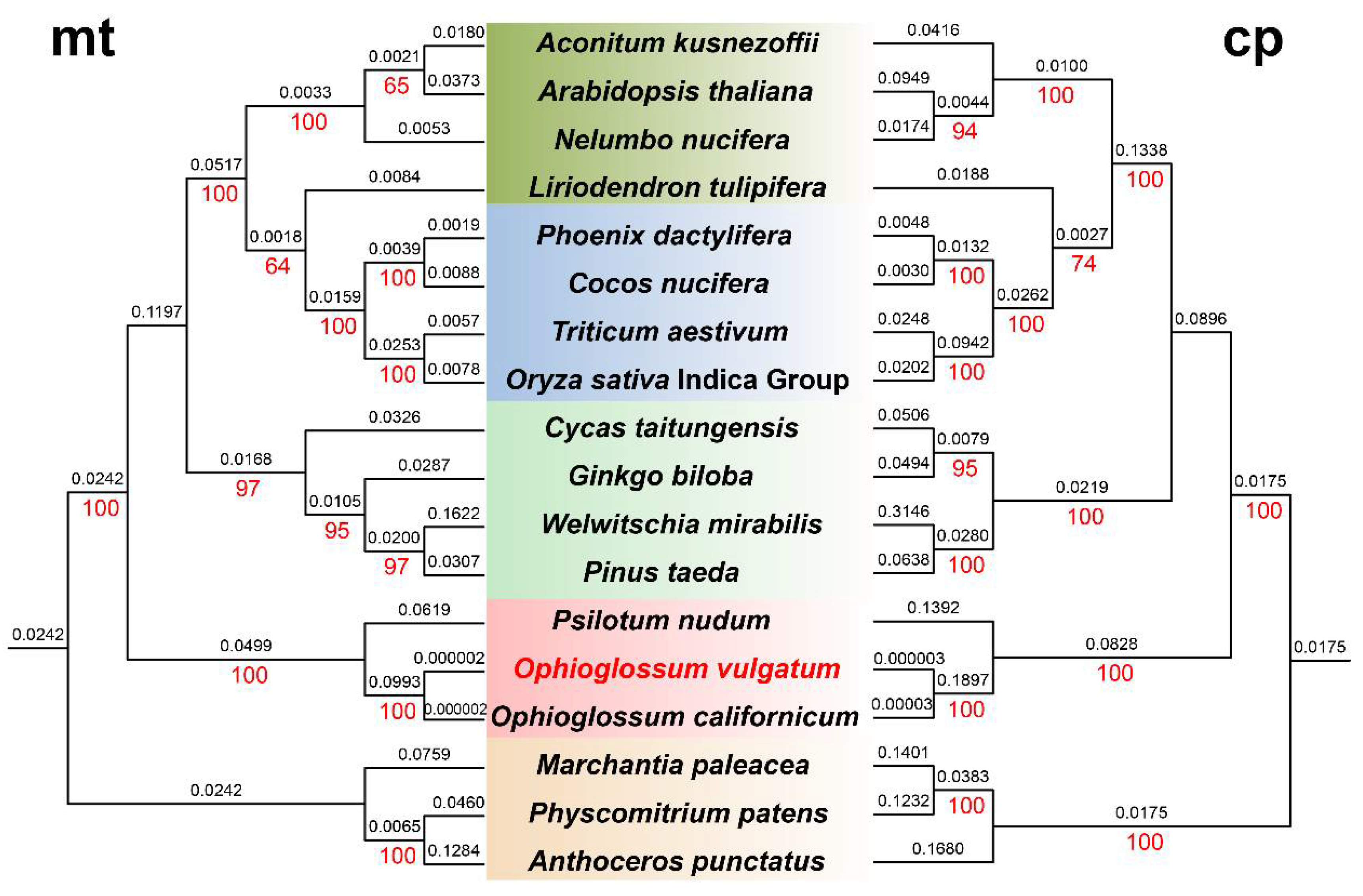
3.3. Comparative Phylogenetic Analysis Based on mt and cp Genomic Sequences
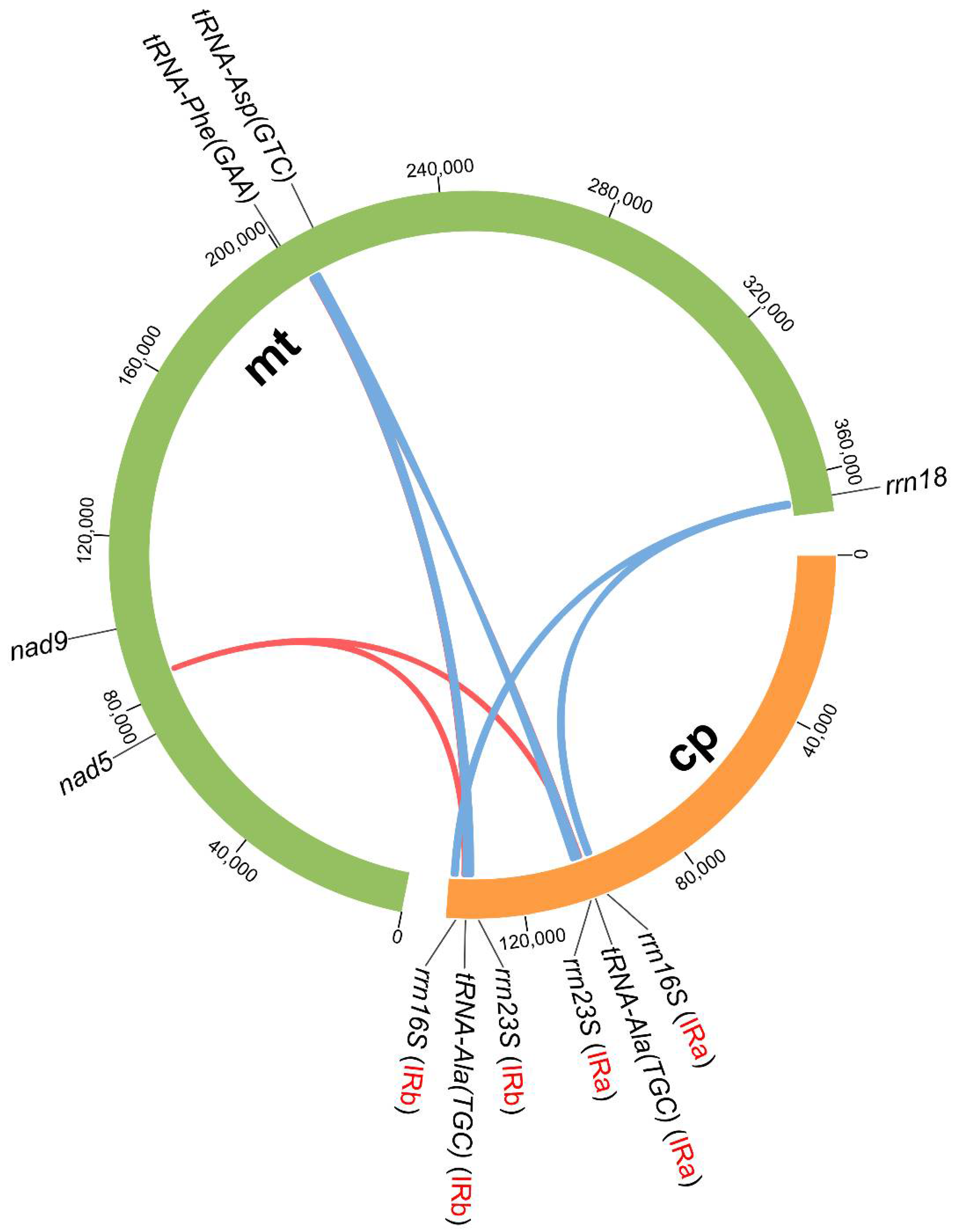
3.4. Intergenomic Fragment Transfers between mt and cp Genomes
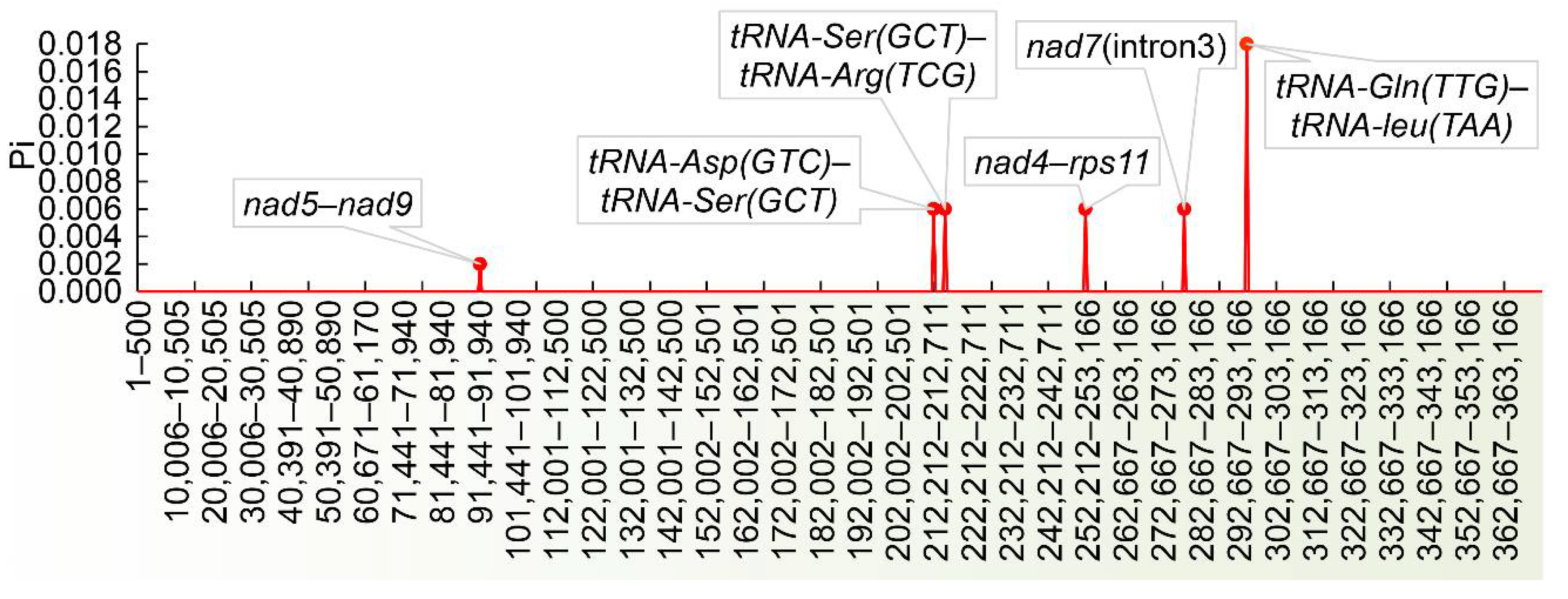
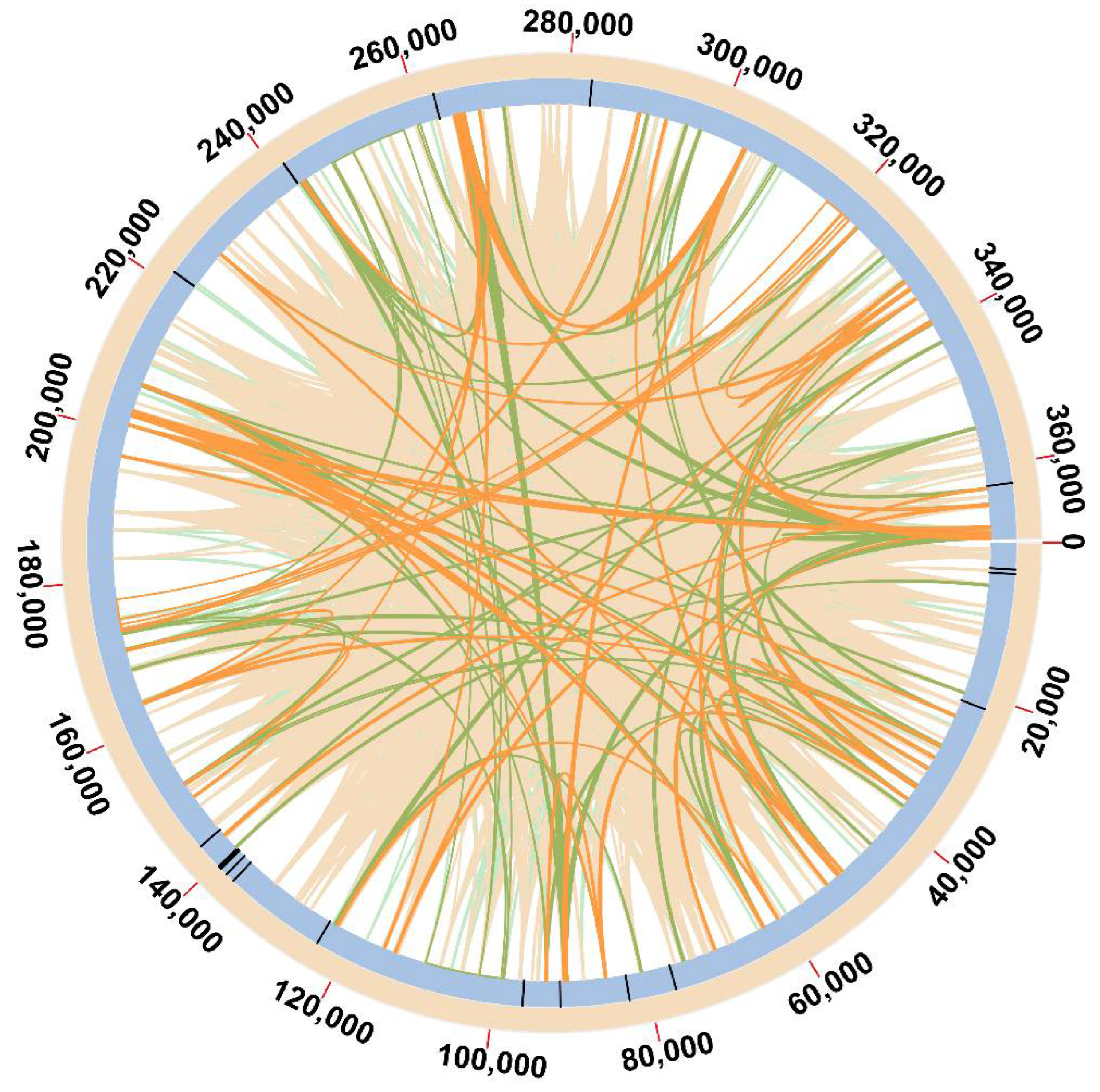
3.5. Analysis of Repetitive Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Handa, H. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): Comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 2003, 31, 5907–5916. [Google Scholar] [PubMed] [Green Version]
- Zhang, X.; Zhang, R.; Hou, S.; Shi, J.; Guo, S. Research progress on mitochondrial genome of higher plant. J. Agric. Sci. Technol. 2011, 13, 23–31. [Google Scholar]
- Hong, Z.; Liao, X.; Ye, Y.; Zhang, N.; Yang, Z.; Zhu, W.; Gao, W.; Sharbrough, J.; Tembrock, L.R.; Xu, D.; et al. A complete mitochondrial genome for fragrant Chinese rosewood (Dalbergia odorifera, Fabaceae) with comparative analyses of genome structure and intergenomic sequence transfers. BMC Genom. 2021, 22, 672. [Google Scholar]
- Morley, S.A.; Nielsen, B.L. Plant mitochondrial DNA. Front. Biosci. 2017, 22, 1023–1032. [Google Scholar]
- Jackman, S.D.; Coombe, L.; Warren, R.L.; Kirk, H.; Trinh, E.; MacLeod, T.; Pleasance, S.; Pandoh, P.; Zhao, Y.; Coope, R.J.; et al. Complete mitochondrial genome of a gymnosperm, sitka spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol. 2020, 12, 1174–1179. [Google Scholar]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar]
- Lonsdale, D.M.; Brears, T.; Hodge, T.P.; Melville, S.E.; Rottmann, W.H. The plant mitochondrial genome: Homologous recombination as a mechanism for generating heterogeneity. Philos. Trans. R. Soc. Lond. B 1988, 319, 149–163. [Google Scholar]
- Guo, W.; Zhu, A.; Fan, W.; Mower, J.P. Complete mitochondrial genomes from the ferns Ophioglossum californicum and Psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 2017, 213, 391–403. [Google Scholar]
- Nugent, J.M.; Palmer, J.D. RNA-mediated transfer of the gene coxII from the mitochondrion to the nucleus during flowering plant evolution. Cell 1991, 66, 473–481. [Google Scholar]
- Kubo, T.; Newton, K.J. Angiosperm mitochondrial genomes and mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar]
- Lei, B.; Li, S.; Liu, G.; Wang, Y.; Su, A.; Hua, J. Evolutionary analysis of mitochondrial genomes in higher plants. Mol. Plant Breed. 2012, 10, 490–500. [Google Scholar]
- Choi, K.S.; Park, S. Complete plastid and mitochondrial genomes of Aeginetia indica reveal intracellular gene transfer (IGT), horizontal gene transfer (HGT), and cytoplasmic male sterility (CMS). Int. J. Mol. Sci. 2021, 22, 6143. [Google Scholar] [PubMed]
- Hao, W.; Palmer, J.D. Fine-scale mergers of chloroplast and mitochondrial genes create functional, transcompartmentally chimeric mitochondrial genes. Proc. Natl. Acad. Sci. USA 2009, 106, 16728–16733. [Google Scholar] [PubMed] [Green Version]
- Smith, D.R. Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol. Evol. 2011, 3, 743–748. [Google Scholar]
- Cusimano, N.; Wicke, S. Massive intracellular gene transfer during plastid genome reduction in nongreen Orobanchaceae. New Phytol. 2016, 210, 680–693. [Google Scholar]
- Wang, S.; Li, D.; Yao, X.; Song, Q.; Wang, Z.; Zhang, Q.; Zhong, C.; Liu, Y.; Huang, H. Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol. Evol. 2019, 11, 1192–1206. [Google Scholar]
- Nock, C.J.; Waters, D.L.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011, 9, 328–333. [Google Scholar]
- Asaf, S.; Khan, A.L.; Khan, A.R.; Waqas, M.; Kang, S.M.; Khan, M.A.; Shahzad, R.; Seo, C.W.; Shin, J.H.; Lee, I.J. Mitochondrial genome analysis of wild rice (Oryza minuta) and its comparison with other related species. PLoS ONE 2016, 11, e152937. [Google Scholar]
- Small, R.L.; Cronn, R.C.; Wendel, J.F. Use of nuclear genes for phylogeny reconstruction in plants. Aust. Syst. Bot. 2004, 17, 145–170. [Google Scholar]
- Duminil, J.; Besnard, G. Utility of the mitochondrial genome in plant taxonomic studies. Methods Mol. Biol. 2021, 2222, 107–118. [Google Scholar] [PubMed]
- Tian, X.; Zheng, J.; Hu, S.; Yu, J. The rice mitochondrial genomes and their variations. Plant Physiol. 2006, 140, 401–410. [Google Scholar] [PubMed] [Green Version]
- Oda, K.; Yamato, K.; Ohta, E.; Nakamura, Y.; Takemura, M.; Nozato, N.; Akashi, K.; Kanegae, T.; Ogura, Y.; Kohchi, T.; et al. Gene organization deduced from the complete sequence of liverwort Marchantia polymorpha mitochondrial DNA: A primitive form of plant mitochondrial genome. J. Mol. Biol. 1992, 223, 5907–5916. [Google Scholar]
- Zhang, L.; Fan, X.P.; Petchsri, S.; Zhou, L.; Pollawatn, R.; Zhang, X.; Zhou, X.M.; Thi, L.N.; Knapp, R.; Chantanaorrapint, S.; et al. Evolutionary relationships of the ancient fern lineage the adder’s tongues (Ophioglossaceae) with description of Sahashia gen. Nov. Cladistics 2020, 36, 380–393. [Google Scholar]
- Clericuzio, M.; Tinello, S.; Burlando, B.; Ranzato, E.; Martinotti, S.; Cornara, L.; La Rocca, A. Flavonoid oligoglycosides from Ophioglossum vulgatum L. having wound healing properties. Planta Med. 2012, 78, 1639–1644. [Google Scholar] [PubMed] [Green Version]
- Zhang, X.; Liu, Q.; Sahashi, N. Ophioglossaceae. In Flora of China; Wu, Z., Raven, P.H., Hong, D., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2013; pp. 73–80. [Google Scholar]
- Hao, J.; Liang, Y.; Zhu, M.; Ping, J.; Feng, P.; Su, Y.; Wang, T. The complete chloroplast genome of Ophioglossum vulgatum L. (Ophioglossaceae) and phylogenetic analysis. Mitochondrial DNA B Resour. 2021, 6, 2730–2731. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [PubMed] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Feng, P.; Ping, J.; Li, J.; Su, Y.; Wang, T. Phylogenetic significance of the characteristics of simple sequence repeats at the genus level based on the complete chloroplast genome sequences of Cyatheaceae. Ecol. Evol. 2021, 11, 14327–14340. [Google Scholar] [CrossRef]
- Adams, K.L.; Palmer, J.D. Evolution of mitochondrial gene content: Gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 2003, 29, 380–395. [Google Scholar] [CrossRef]
- Giegé, P.; Grienenberger, J.M.; Bonnard, G. Cytochrome c biogenesis in mitochondria. Mitochondrion 2008, 8, 61–73. [Google Scholar] [CrossRef]
- Miyata, S.; Nakazono, M.; Hirai, A. Transcription of plastid-derived tRNA genes in rice mitochondria. Curr. Genet. 1998, 34, 216–220. [Google Scholar] [CrossRef]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Zhao, C.; Chen, F.; Liu, Y.; Zhang, S.; Wu, H.; Zhang, L.; Liu, Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genom. 2018, 19, 614. [Google Scholar] [CrossRef] [PubMed]
- Van de Paer, C.; Bouchez, O.; Besnard, G. Prospects on the evolutionary mitogenomics of plants: A case study on the olive family (Oleaceae). Mol. Ecol. Resour. 2018, 18, 407–423. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.S.; McCauley, D.E. Linkage disequilibrium and phylogenetic congruence between chloroplast and mitochondrial haplotypes in Silene vulgaris. Proc. Biol. Sci. 2000, 267, 1801–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Names | Family | Group | Genbank Accessions (mt) | Genbank Accessions (cp) |
|---|---|---|---|---|
| Marchantia paleacea | Marchantiaceae | Bryophyte | NC_001660 | NC_001319 |
| Physcomitrium patens | Funariaceae | Bryophyte | NC_007945 | KY126308 |
| Anthoceros punctatus | Anthocerotaceae | Bryophyte | NC_049003 | MN544310 |
| Ophioglossum californicum | Ophioglossaceae | Fern | KX171637 | NC_020147 |
| O. vulgatum | Ophioglossaceae | Fern | OL800577 | MZ066610 |
| Psilotum nudum | Psilotaceae | Fern | KX171638, KX171639 | NC_003386 |
| Welwitschia mirabilis | Welwitschiaceae | Gymnosperm | NC_029130 | NC_010654 |
| Pinus taeda | Pinaceae | Gymnosperm | NC_039746 | NC_021440 |
| Cycas taitungensis | Cycadaceae | Gymnosperm | NC_010303 | NC_009618 |
| Ginkgo biloba | Ginkgoaceae | Gymnosperm | NC_027976 | NC_016986 |
| Oryza sativa Indica Group | Poaceae | Angiosperm (monocot) | NC_007886 | NC_008155 |
| Triticum aestivum | Poaceae | Angiosperm (monocot) | GU985444 | NC_002762 |
| Phoenix dactylifera | Arecaceae | Angiosperm (monocot) | NC_016740 | NC_013991 |
| Cocos nucifera | Arecaceae | Angiosperm (monocot) | NC_031696 | NC_022417 |
| Liriodendron tulipifera | Magnoliaceae | Angiosperm (dicot) | NC_021152 | NC_008326 |
| Aconitum kusnezoffii | Ranunculaceae | Angiosperm (dicot) | NC_053920 | KT820671 |
| Nelumbo nucifera | Nelumbonaceae | Angiosperm (dicot) | NC_030753 | NC_025339 |
| Arabidopsis thaliana | Brassicaceae | Angiosperm (dicot) | Y08501 | NC_000932 |
| Gene Type | Gene Name |
|---|---|
| ATPase subunits | atp1, atp4, atp6, atp8, atp9 |
| Apocytochrome b | cob |
| Cytochrome c oxidase subunits | cox1, cox2, cox3 |
| Ribosomal proteins | rpl2, rpl5, rpl6, rpl16, rps1, rps2, rps3, rps4, rps7, rps10, rps11, rps12, rps13, rps14, rps19 |
| Maturase | matR |
| Sec-independent protein translocase protein | mttB |
| NADH dehydrogenase subunits | nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9 |
| Succinate dehydrogenase cytochrome subunits | sdh3, sdh4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, J.; Liang, Y.; Su, Y.; Wang, T. The Complete Mitochondrial Genome of Ophioglossum vulgatum L. Is with Highly Repetitive Sequences: Intergenomic Fragment Transfer and Phylogenetic Analysis. Genes 2022, 13, 1287. https://doi.org/10.3390/genes13071287
Hao J, Liang Y, Su Y, Wang T. The Complete Mitochondrial Genome of Ophioglossum vulgatum L. Is with Highly Repetitive Sequences: Intergenomic Fragment Transfer and Phylogenetic Analysis. Genes. 2022; 13(7):1287. https://doi.org/10.3390/genes13071287
Chicago/Turabian StyleHao, Jing, Yingyi Liang, Yingjuan Su, and Ting Wang. 2022. "The Complete Mitochondrial Genome of Ophioglossum vulgatum L. Is with Highly Repetitive Sequences: Intergenomic Fragment Transfer and Phylogenetic Analysis" Genes 13, no. 7: 1287. https://doi.org/10.3390/genes13071287
APA StyleHao, J., Liang, Y., Su, Y., & Wang, T. (2022). The Complete Mitochondrial Genome of Ophioglossum vulgatum L. Is with Highly Repetitive Sequences: Intergenomic Fragment Transfer and Phylogenetic Analysis. Genes, 13(7), 1287. https://doi.org/10.3390/genes13071287