
Genetic Profile of Left Ventricular Noncompaction Cardiomyopathy in Children—A Single Reference Center Experience

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Selection and Clinical Data Collection
2.2. Molecular Studies and Data Analysis
2.3. Ethics Statement
3. Results
3.1. Clinical Characteristics of the Study Group
3.2. Family History
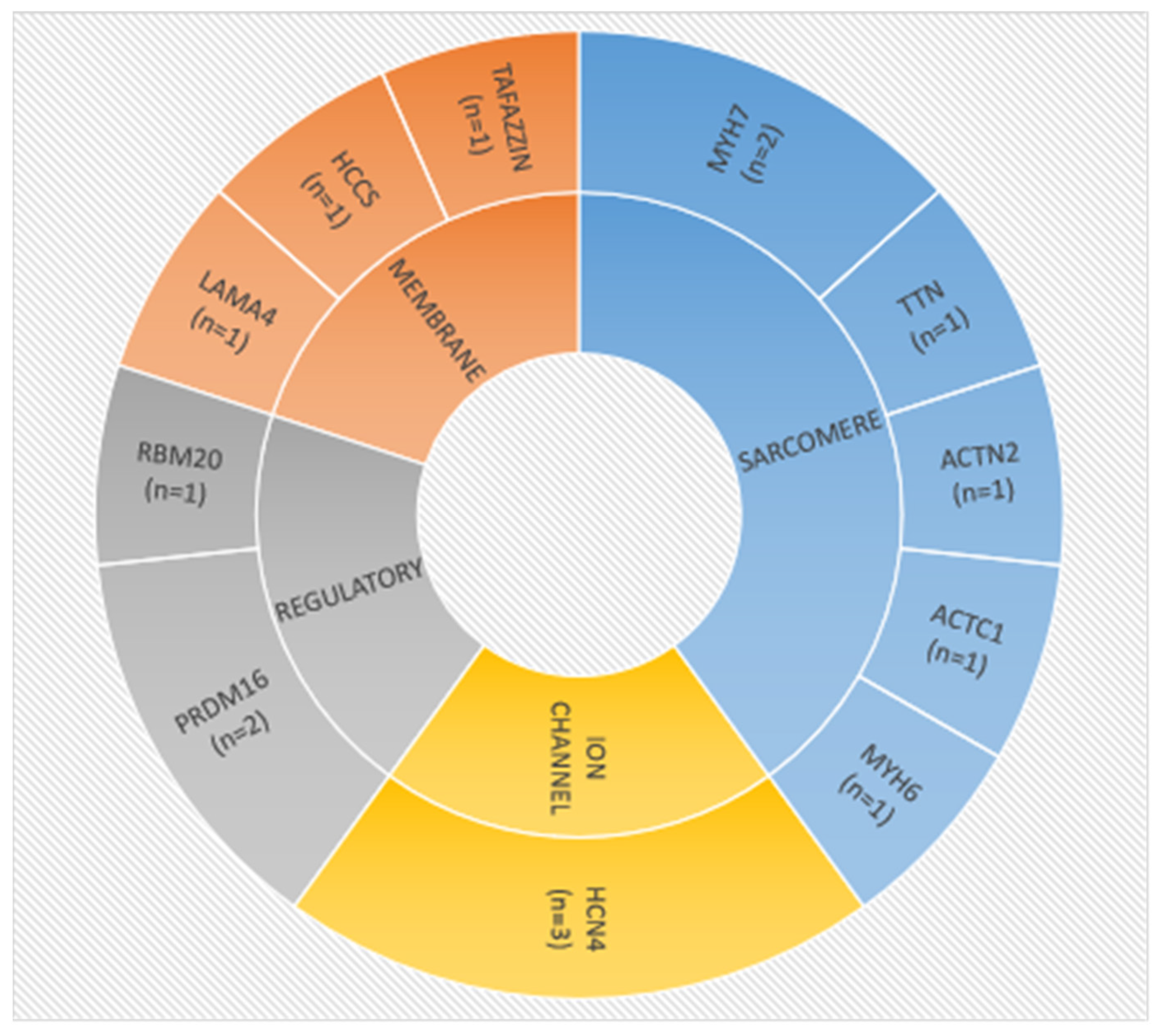
3.3. Molecular Characteristics
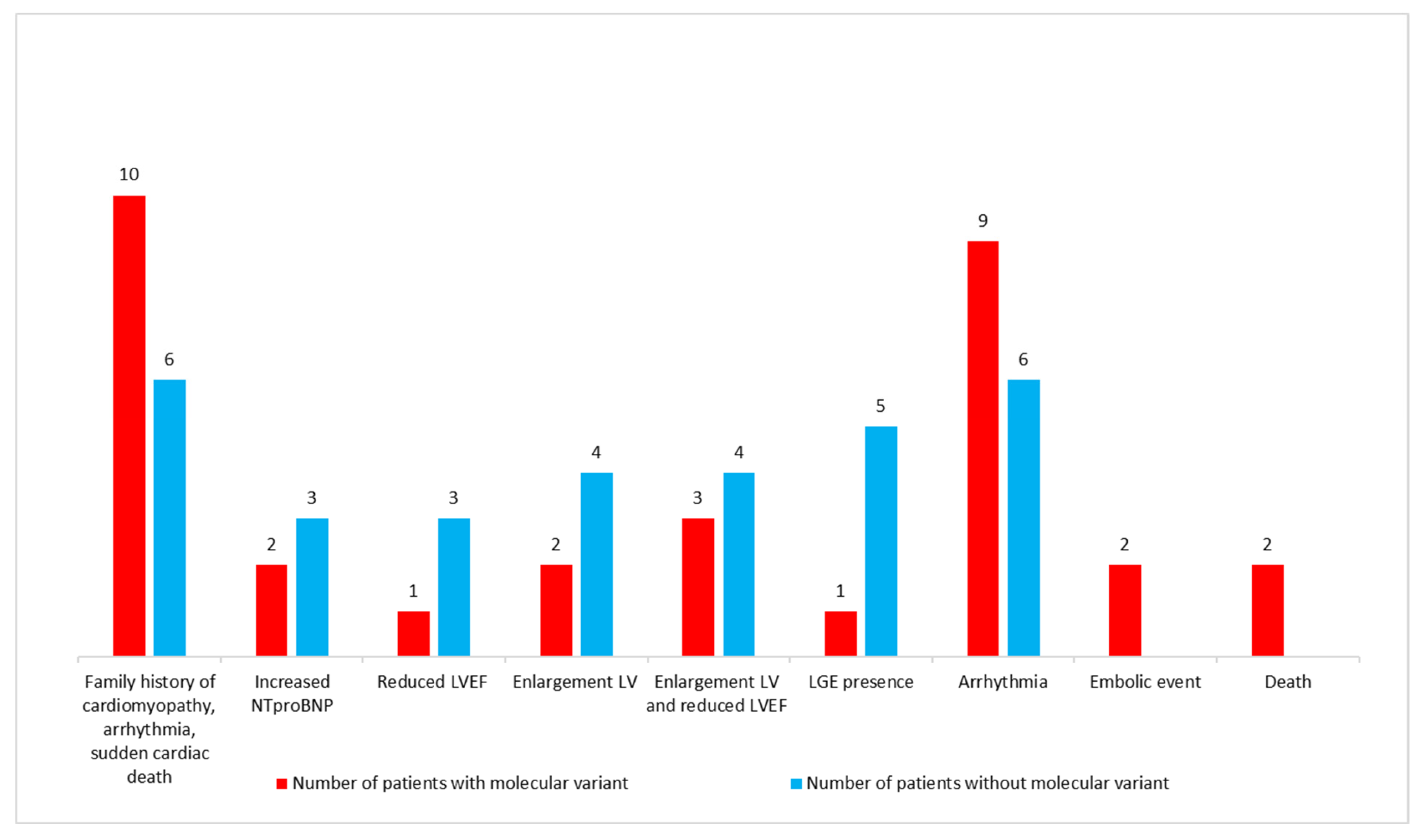
3.4. Characteristics of Patients with Putative Disease-Causing Variants
3.5. Patients with Unknown Molecular Etiology
4. Discussion
5. Study Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ichida, F. Left ventricular noncompaction-Risk stratification and genetic consideration. J. Cardiol. 2020, 75, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisz, S.H.; Limongelli, G.; Pacileo, G.; Calabro, P.; Russo, M.G.; Calabro, R.; Vatta, M. Left ventricular non compaction in children. Congenit. Heart Dis. 2010, 5, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Paterick, T.E.; Tajik, A.J. Left ventricular noncompaction: A diagnostically challenging cardiomyopathy. Circ. J. 2012, 76, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferies, J.L.; Wilkinson, J.D.; Sleeper, L.A.; Colan, S.D.; Lu, M.; Pahl, E.; Kantor, P.F.; Everitt, M.D.; Webber, S.A.; Kaufman, B.D.; et al. Pediatric Cardiomyopathy Registry Investigators. Cardiomyopathy Phenotypes and Outcomes for Children with Left Ventricular Myocardial Noncompaction: Results from the Pediatric Cardiomyopathy Registry. J. Card. Fail. 2015, 21, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Miszalski-Jamka, K.; Jefferies, J.L.; Mazur, W.; Głowacki, J.; Hu, J.; Lazar, M.; Gibbs, R.A.; Liczko, J.; Kłyś, J.; Venner, E.; et al. Novel Genetic Triggers and Genotype-Phenotype Correlations in Patients with Left Ventricular Noncompaction. Circ. Cardiovasc. Genet. 2017, 10, e001763. [Google Scholar] [CrossRef] [Green Version]
- Jenni, R.; Oechslin, E.; Schneider, J.; Attenhofer Jost, C.; Kaufmann, P.A. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: A step towards classification as a distinct cardiomyopathy. Heart 2001, 86, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left ventricular non-compaction: Insights from cardiovascular magnetic resonance imaging. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, H.; Shou, W. Potential Common Pathogenic Pathways for the Left Ventricular Noncompaction Cardiomyopathy (LVNC). Pediatr Cardiol. 2018, 39, 1099–1106. [Google Scholar] [CrossRef]
- Wu, T.; Liang, Z.; Zhang, Z.; Liu, C.; Zhang, L.; Gu, Y.; Peterson, K.L.; Evans, S.M.; Fu, X.D.; Chen, J. PRDM16 Is a Compact Myocardium-Enriched Transcription Factor Required to Maintain Compact Myocardial Cardiomyocyte Identity in Left Ventricle. Circulation 2022, 145, 586–602. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lorts, A.; Jefferies, J.L. Left ventricular non-compaction cardiomyopathy. Lancet 2015, 386, 813–825. [Google Scholar] [CrossRef]
- Finsterer, J.; Stöllberger, C. Left Ventricular Noncompaction Syndrome: Genetic Insights and Therapeutic Perspectives. Curr. Cardiol. Rep. 2020, 22, 84. [Google Scholar] [CrossRef] [PubMed]
- Bleyl, S.B.; Mumford, B.R.; Thompson, V.; Carey, J.C.; Pysher, T.J.; Chin, T.K.; Ward, K. Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am. J. Hum. Genet. 1997, 61, 868–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszkowska, A.; Mirecka-Rola, A.; Piekutowska-Abramczuk, D.; Ciara, E.; Mazurkiewicz, Ł.; Bieganowska, K.; Ziółkowska, L. Spectrum of Clinical Features and Genetic Profile of Left Ventricular Noncompaction Cardiomyopathy in Children. Cardiogenetics 2021, 11, 191–203. [Google Scholar] [CrossRef]
- Paszkowska, A.; Piekutowska-Abramczuk, D.; Ciara, E.; Mirecka-Rola, A.; Brzezinska, M.; Wicher, D.; Kostrzewa, G.; Sarnecki, J.; Ziółkowska, L. Clinical Presentation of Left Ventricular Noncompaction Cardiomyopathy and Bradycardia in Three Families Carrying HCN4 Pathogenic Variants. Genes 2022, 13, 477. [Google Scholar] [CrossRef]
- Fleming, S.; Thompson, M.; Stevens, R.; Heneghan, C.; Plüddemann, A.; Maconochie, I.; Tarassenko, L.; Mant, D. Normal ranges of heart rate and respiratory rate in children from birth to 18 years of age: A systematic review of observational studies. Lancet. 2011, 377, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Baruteau, A.E.; Perry, J.C.; Sanatani, S.; Horie, M.; Dubin, A.M. Evaluation and management of bradycardia in neonates and children. Eur. J. Pediatr. 2016, 175, 151–161. [Google Scholar] [CrossRef]
- Kampmann, C.; Wiethoff, C.M.; Wenzel, A.; Stolz, G.; Betancor, M.; Wippermann, C.F.; Huth, R.G.; Habermehl, P.; Knuf, M.; Emschermann, T.; et al. Normal values of M mode echocardiographic measurements of more than 2000 healthy infants and children in central Europe. Heart 2000, 83, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Chubb, H.; Simpson, J.M. The use Z-score in paediatric cardiology. Ann. Pediatr. Cardiol. 2012, 5, 179–184. [Google Scholar]
- Paszkowska, A.; Sarnecki, J.; Mirecka-Rola, A.; Kowalczyk-Domagała, M.; Mazurkiewicz, Ł.; Ziółkowska, L. Imaging Features of Pediatric Left Ventricular Noncompaction Cardiomyopathy in Echocardiography and Cardiovascular Magnetic Resonance. J. Cardiovasc. Dev. Dis. 2022, 9, 77. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-wide copy number detection and visualization from targeted sequencing. PLoS Comput. Biol. 2014, 12, e1004873. [Google Scholar] [CrossRef]
- Ciara, E.; Rokicki, D.; Lazniewski, M.; Mierzewska, H.; Jurkiewicz, E.; Bekiesińska-Figatowska, M.; Piekutowska-Abramczuk, D.; Iwanicka-Pronicka, K.; Szymańska, E.; Stawiński, P.; et al. Clinical and molecular characteristics of newly reported mitochondrial disease entity caused by biallelic PARS2 mutations. J. Hum. Genet. 2018, 63, 473–485. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients with Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- van Waning, J.I.; Caliskan, K.; Hoedemaekers, Y.M.; van Spaendonck-Zwarts, K.Y.; Baas, A.F.; Boekholdt, S.M.; van Melle, J.P.; Teske, A.J.; Asselbergs, F.W.; Backx, A.P.C.M.; et al. Genetics, clinical features, and long-term outcome of noncompaction cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 711–722. [Google Scholar] [CrossRef]
- Ross, S.B.; Singer, E.S.; Driscoll, E.; Nowak, N.; Yeates, L.; Puranik, R.; Sy, R.W.; Rajagopalan, S.; Barratt, A.; Ingles, J.; et al. Genetic architecture of left ventricular noncompaction in adults. Hum. Genome Var. 2020, 7, 33. [Google Scholar] [CrossRef]
- Miller, E.M.; Hinton, R.B.; Czosek, R.; Lorts, A.; Parrott, A.; Shikany, A.R.; Ittenbach, R.F.; Ware, S.M. Genetic Testing in Pediatric Left Ventricular Noncompaction. Circ. Cardiovasc. Genet. 2017, 10, e001735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzarotto, F.; Hawley, M.H.; Beltrami, M.; Beekman, L.; de Marvao, A.; McGurk, K.A.; Statton, B.; Boschi, B.; Girolami, F.; Roberts, A.M.; et al. Systematic large-scale assessment of the genetic architecture of left ventricular noncompaction reveals diverse etiologies. Genet. Med. 2021, 23, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, C.; Liu, N.; Bai, H.; Hou, C.; Wang, J.; Song, L.; Pu, J. Genotype-Positive Status Is Associated with Poor Prognoses in Patients With Left Ventricular Noncompaction Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e009910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Waning, J.I.; Moesker, J.; Heijsman, D.; Boersma, E.; Majoor-Krakauer, D. Systematic Review of Genotype-Phenotype Correlations in Noncompaction Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e012993. [Google Scholar] [CrossRef]
- Hirono, K.; Hata, Y.; Miyao, N.; Okabe, M.; Takarada, S.; Nakaoka, H.; Ibuki, K.; Ozawa, S.; Origasa, H.; Nishida, N.; et al. LVNC study collaborates. Increased Burden of Ion Channel Gene Variants Is Related to Distinct Phenotypes in Pediatric Patients with Left Ventricular Noncompaction. Circ. Genom. Precis. Med. 2020, 13, e002940. [Google Scholar] [CrossRef]
- Hirono, K.; Hata, Y.; Ozawa, S.W.; Toda, T.; Momoi, N.; Fukuda, Y.; Inuzuka, R.; Nagamine, H.; Sakaguchi, H.; Kurosaki, K.; et al. A burden of sarcomere gene variants in fetal-onset patients with left ventricular noncompaction. Int. J. Cardiol. 2021, 328, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Janin, A.; de Tauriac, O.; Roux, A.; Dauphin, C. HCN4 mutation as a molecular explanation on patients with bradycardia and non-compaction cardiomyopathy. Eur. J. Med. Genet. 2015, 58, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Ader, F.; Roux, M.; Donal, E.; Eicher, J.C.; Aoutil, N.; Huttin, O.; Selton-Suty, C.; Coisne, D.; Jondeau, G.; et al. Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin. Genet. 2019, 95, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Cambon-Viala, M.; Gerard, H.; Nguyen, K.; Richard, P.; Ader, F.; Pruny, J.F.; Donal, E.; Eicher, J.C.; Huttin, O.; Selton-Suty, C.; et al. Phenotype/Genotype Relationship in Left Ventricular Noncompaction: Ion Channel Gene Mutations Are Associated with Preserved Left Ventricular Systolic Function and Biventricular Noncompaction: Phenotype/Genotype of Noncompaction. J. Card. Fail. 2021, 27, 677–681. [Google Scholar] [CrossRef]
- Towbin, J.A. Ion channel dysfunction associated with arrhythmia, ventricular noncompaction, and mitral valve prolapse: A new overlapping phenotype. J. Am. Coll. Cardiol. 2014, 64, 768–771. [Google Scholar] [CrossRef] [Green Version]
- Servatius, H.; Porro, A.; Pless, S.A.; Schaller, A.; Asatryan, B.; Tanner, H.; de Marchi, S.F.; Roten, L.; Seiler, J.; Haeberlin, A.; et al. Phenotypic Spectrum of HCN4 Mutations: A Clinical Case. Circ. Genom. Precis. Med. 2018, 11, e002033. [Google Scholar] [CrossRef] [Green Version]
- Takasaki, A.; Hirono, K.; Hata, Y.; Wang, C.; Takeda, M.; Yamashita, J.K.; Chang, B.; Nakaoka, H.; Okabe, M.; Miyao, N.; et al. Sarcomere gene variants act as a genetic trigger underlying the development of left ventricular noncompaction. Pediatr. Res. 2018, 84, 733–742. [Google Scholar] [CrossRef]
- van Waning, J.I.; Caliskan, K.; Michels, M.; Schinkel, A.F.L.; Hirsch, A.; Dalinghaus, M.; Hoedemaekers, Y.M.; Wessels, M.W.; IJpma, A.S.; Hofstra, R.M.W.; et al. Cardiac Phenotypes, Genetics, and Risks in Familial Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 73, 1601–1611. [Google Scholar] [CrossRef]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef]
- Lennermann, D.; Backs, J.; van den Hoogenhof, M.M.G. New Insights in RBM20 Cardiomyopathy. Curr. Heart Fail. Rep. 2020, 17, 234–246. [Google Scholar] [CrossRef]
- Adwani, S.S.; Whitehead, B.F.; Rees, P.G.; Morris, A.; Turnball, D.M.; Elliott, M.J.; de Level, M.R. Heart transplantation for Barth syndrome. Pediatr. Cardiol. 1997, 18, 143–145. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Patient | Age (Yers) | Gender | Syncope | Embolic Event | Dysmorphic Features | Heart Failure Profile | Arrhythmias and Atrioventricular Conduction Disorders | EPS/RFA | Pacemaker | LVAD | Death | Family History | NC/C (Echo) | NC/C (CMR) | LGE % | Affected Gene | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NYHA/Ross Class | NTproBNP pg/mL | LVEF (ECHO) | LVDd mm (z-Score) ECHO | LVEF (CMR) | LVEDV mL/BSA (z-Score) | CMP | Arrhythmias | SCD | |||||||||||||||
| P1 | 1 | F | 0 | 0 | 0 | II | 256.50 | 63 | 32.2 (2) | N/A | N/A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6.3 | N/A | N/A | LAMA4 |
| P2 | 2 | F | 0 | 1 | 0 | IV | N/A | 59 | 28.4 (0.5) | N/A | N/A | Brady | 0 | 0 | 0 | 1 | LVNC (sister) | Brady (sister, father’s father) | 0 | 3.34 | N/A | N/A | HCN4 |
| P3 | 6 | F | 0 | 0 | 0 | II | 110.80 | 64 | 35.6 (−0.5) | 71 | 73 (0.7) | Brady | 0 | 0 | 0 | 0 | LVNC (sister) | Brady (sister, father’s father) | 1 | 2.35 | 4.6 | 0 | HCN4 |
| P4 | 11 | F | 0 | 0 | 0 | I | <5.0 | 70 | 46.3 (0.9) | 71.3 | 69.6 (1.2) | Brady | 0 | 0 | 0 | 0 | LVNC (mother, 2 cousins) | 0 | 0 | 2.22 | 3.0 | 0 | HCN4 |
| P5 | 17 | M | 0 | 0 | 0 | II | 32.95 | 71.5 | 41.1 (0.8) | 63.7 | 125.8 (−1) | Brady | 0 | 0 | 0 | 0 | LVNC (sister), HCM (father) | Brady (2 sisters) | 0 | 2.4 | 3.73 | 4.7 | HCN4 |
| P6 | 5 | F | 1 | 0 | 0 | II | N/A | 61 | 38 (−0.1) | N/A | N/A | Brady | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2.85 | N/A | N/A | MYH7 |
| P7 | 12 | F | 1 | 0 | 0 | II | 68.54 | 50 | 52.2 (2.1) | 54.5 | 103.5 (3) | Brady | 0 | 0 | 0 | 0 | LVNC (mother) | 0 | 0 | 2.6 | 3.38 | 0 | MYH7 |
| P8 | 6 | F | 0 | 1 | 0 | II | 38.69 | 61 | 37 (0.4) | 56 | 71 (0.9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.26 | 2.3 | 0 | PRDM16 |
| P9 | 11 | F | 0 | 0 | 0 | II | 331.20 | 52 | 54.2 (3.6) | 52.4 | 80.9 (1.4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.6 | 3.52 | 0 | PRDM16 |
| P10 | 6 | M | 0 | 0 | 1 | IV | 27057.0 | 28 | 36 (3.5) | N/A | N/A | nsVT | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 3.9 | N/A | N/A | TAFFAZIN |
| P11 | 6 | F | 0 | 0 | 0 | I | 99.89 | 64 | 37.3 (−0.4) | 68 | 68.6 (0.9) | 0 | 0 | 0 | 0 | 0 | HCM (father, father’s sister and father’s mother) | 0 | 1 | 2.7 | 3.09 | 0 | MYH6 |
| P12 | 11 | F | 0 | 0 | 0 | II | 62.91 | 56 | 34 (0.3) | 68.7 | 66.7 (−3.6) | 0 | 0 | 0 | 0 | 0 | 0 | SVT (father’s father) | 0 | 4.4 | 3.76 | 0 | RBM20 |
| P13 | 13 | F | 0 | 0 | 0 | II | 33.30 | 57 | 57 (3.6) | 55.9 | 90.9 (1.8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3.9 | 5.74 | 0 | ACTN2 |
| P14 | 15 | F | 0 | 0 | 1 | II | 13.32 | 63 | 45 (−0.1) | 74.2 | 77 (0.5) | WPW, SVT | 1 | 0 | 0 | 0 | 0 | WPW (father’s father) | 0 | 2.22 | 2.46 | 0 | HCCS |
| P15 | 16 | M | 0 | 0 | 0 | II | 131.70 | 59 | 52.3 (3) | 55.6 | 79.5 (−1.5) | nsVT | 0 | 0 | 0 | 0 | LVNC (father) | 0 | 0 | 2.13 | 2.43 | 0 | TTN |
| P16 | 17 | M | 0 | 0 | 0 | II | 32.73 | 70 | 52.6 (−0.2) | 62.2 | 78.8 (1.1) | 0 | 0 | 0 | 0 | 0 | LVNC (mother, sister, mother’s father) | 0 | 0 | 3.3 | 2.26 | 0 | ACTC1 |
| P17 | 0.2 | F | 0 | 0 | 0 | II | 577.80 | 54 | 23.5 (2.8) | N/A | N/A | 0 | 0 | 0 | 0 | 0 | LVNC (father) | 0 | 0 | 4.8 | N/A | N/A | 0 |
| P18 | 5 | F | 0 | 0 | 0 | II | 1325.0 | 50 | 42 (2.5) | 61.6 | 76.3 (1.2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4.0 | 4.22 | 0 | 0 |
| P19 | 5 | M | 0 | 0 | 0 | II | 65.34 | 63 | 36.4 (0.8) | 53 | 78 (1.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3.4 | 2.65 | 0 | 0 |
| P20 | 6 | M | 0 | 0 | 0 | II | 98.64 | 57 | 37.1 (1) | 67.6 | 104.3 (2.1) | WPW | 0 | 0 | 0 | 0 | 0 | WPW (father’s sister) | 1 | 2.32 | 2.36 | 0 | 0 |
| P21 | 6 | M | 0 | 0 | 0 | II | 118.80 | 53 | 36.6 (0.8) | 54.2 | 54.4 (0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.8 | 2.63 | 9.5 | 0 |
| P22 | 8 | M | 0 | 0 | 0 | II | 349.40 | 55 | 37.8 (−0.5) | 48.1 | 56.5 (−0.7) | 0 | 0 | 0 | 0 | 0 | LVNC (mother) | 0 | 0 | 2.13 | 2.39 | 0 | 0 |
| P23 | 10 | F | 0 | 0 | 0 | II | 63.01 | 51 | 32.8 (−1.7) | 71.7 | 61.1 (−1.8) | SVT | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2.2 | 3.27 | 7.8 | 0 |
| P24 | 11 | M | 0 | 0 | 0 | II | 38.88 | 63 | 56 (4.6) | 63 | 88.7 (0.9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5.14 | 5.07 | 0 | 0 |
| P25 | 13 | F | 0 | 0 | 0 | II | 67.14 | 56 | 51 (2.6) | 58.4 | 83.2 (0.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.22 | 3.45 | 0 | 0 |
| P26 | 13 | M | 0 | 0 | 0 | I | 81.78 | 64 | 51.5 (1.9) | 62.9 | 85.8 (0.9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.9 | 2.60 | 0 | 0 |
| P27 | 13 | F | 1 | 0 | 0 | II | 30.07 | 60 | 35.7 (−2) | 72.9 | 60.2 (−1.1) | 0 | 0 | 0 | 0 | 0 | 0 | AVNRT (mother) | 0 | 2.85 | 2.56 | 0 | 0 |
| P28 | 15 | M | 0 | 0 | 0 | II | 73.13 | 53 | 51.8 (2.8) | N/A | N/A | AV block | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3.1 | N/A | 0 | 0 |
| P29 | 16 | M | 0 | 0 | 0 | II | 8.76 | 65 | 58 (2.8) | 57.3 | 130 (3) | Brady, AV block | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3.9 | 2.45 | 3 | 0 |
| P30 | 11 | M | 0 | 0 | 0 | II | 19.04 | 62 | 47.3 (2.3) | 70.2 | 114.1 (1.2) | Brady, AV block | 0 | 0 | 0 | 0 | LVNC (sister); DCM (father) | Brady (sister, father, father’s father) | 0 | 2.13 | 3.27 | 8 | 0 |
| P31 | 16 | F | 0 | 0 | 0 | II | 91.10 | 55 | 59.5 (2.9) | 54.2 | 122.1 (3.7) | Brady, AV block | 0 | 0 | 0 | 0 | LVNC (brother); DCM (father) | Brady (brother, father, father’s father) | 0 | 2.06 | 3.91 | 6.7 | 0 |
| Gene | Chromosome | Transcript | Nucleotide Change | Protein Effect | Variant Type (Consequence) | Pathogenicity | Patient | Family Segregation | CMP Family History | References |
|---|---|---|---|---|---|---|---|---|---|---|
| ACTC1 | 15 | NM_005159.5 | c.329C>T | p.(Ala110Val) | missense | VUS | P16 | NA | LVNC (mother, sister, mother’s father) | [5] |
| ACTN2 | 1 | NM_001103.4 | c.1163G>A | p.(Trp388*) | nonsense (LOF) | P | P13 | NA | no | this study |
| HCCS | X | NM_005333.5 | c.789G>A | p.(Trp263*) | nonsense (LOF) | P | P14 | NA | no | this study |
| HCN4 | 15 | NM_005477.3 | c.1444G>A | p.(Gly482Arg) | missense | P | P2, P3 (siblings) | paternal | LVNC (sister) | [14] |
| HCN4 | 15 | NM_005477.3 | c.1454C>T | p.(Ala485Val) | missense | P | P4 | maternal | LVNC (mother, 2 cousins) | [14] |
| HCN4 | 15 | NM_005477.3 | c.1438G>C | p.(Gly480Arg) | missense | LP | P5 | paternal | LVNC (sister); HCM (father) | [14] |
| LAMA4 | 6 | NM_002290.5 | c.719-1G>T | p.? | splicing (LOF) | LP | P1 | NA | no | this study |
| MYH6 | 14 | NM_002471.4 | c.4850A>C | p.(Lys1617Thr) | missense | VUS | P11 | NA | HCM (father, father’s sister and father’s mother) | [5] |
| MYH7 | 14 | NM_000257.4 | c.323G>A | p.(Arg108His) | missense | P | P6 | de novo | no | this study |
| MYH7 | 14 | NM_000257.4 | c.3973-2A>C | p.? | splicing (LOF) | P | P7 | maternal | LVNC (mother) | this study |
| PRDM16 | 1 | NM_022114.4 | c.1336G>T | p.(Glu446*) | nonsense (LOF) | P | P8 | de novo | no | this study |
| PRDM16 | 1 | NM_022114.4 | c.1286_1289delinsTTGCACTT | p.(Gly429Valfs*176) | indel (LOF) | P | P9 | de novo | no | this study |
| RBM20 | 10 | NM_001134363.3 | c.1232C>T | p.(Pro411Leu) | missense | VUS | P12 | maternal | no | this study |
| RBM20 | 10 | NM_001134363.3 | c.1958C>T | p.(Thr653Ile) | missense | VUS | P12 | paternal | no | [23] |
| TAFAZZIN | X | NM_000116.5 | c.(460+1_461-1)_(699+1_700-1)del | p.? | gross deletion (LOF) | P | P10 | NA | no | this study |
| TTN | 2 | NM_001267550.2 | c.44281+1G>T | p.? | splicing (LOF) | LP | P15 | paternal | LVNC (father) | this study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piekutowska-Abramczuk, D.; Paszkowska, A.; Ciara, E.; Frączak, K.; Mirecka-Rola, A.; Wicher, D.; Pollak, A.; Rutkowska, K.; Sarnecki, J.; Ziółkowska, L. Genetic Profile of Left Ventricular Noncompaction Cardiomyopathy in Children—A Single Reference Center Experience. Genes 2022, 13, 1334. https://doi.org/10.3390/genes13081334
Piekutowska-Abramczuk D, Paszkowska A, Ciara E, Frączak K, Mirecka-Rola A, Wicher D, Pollak A, Rutkowska K, Sarnecki J, Ziółkowska L. Genetic Profile of Left Ventricular Noncompaction Cardiomyopathy in Children—A Single Reference Center Experience. Genes. 2022; 13(8):1334. https://doi.org/10.3390/genes13081334
Chicago/Turabian StylePiekutowska-Abramczuk, Dorota, Agata Paszkowska, Elżbieta Ciara, Kamila Frączak, Alicja Mirecka-Rola, Dorota Wicher, Agnieszka Pollak, Karolina Rutkowska, Jędrzej Sarnecki, and Lidia Ziółkowska. 2022. "Genetic Profile of Left Ventricular Noncompaction Cardiomyopathy in Children—A Single Reference Center Experience" Genes 13, no. 8: 1334. https://doi.org/10.3390/genes13081334