Prenatal Diagnosis of Jeune Syndrome Caused by Compound Heterozygous Variants in DYNC2H1 Gene—Case Report with Rapid WES Procedure and Differential Diagnosis of Lethal Skeletal Dysplasias

 , , , , and
, , , , and
Abstract
:1. Introduction
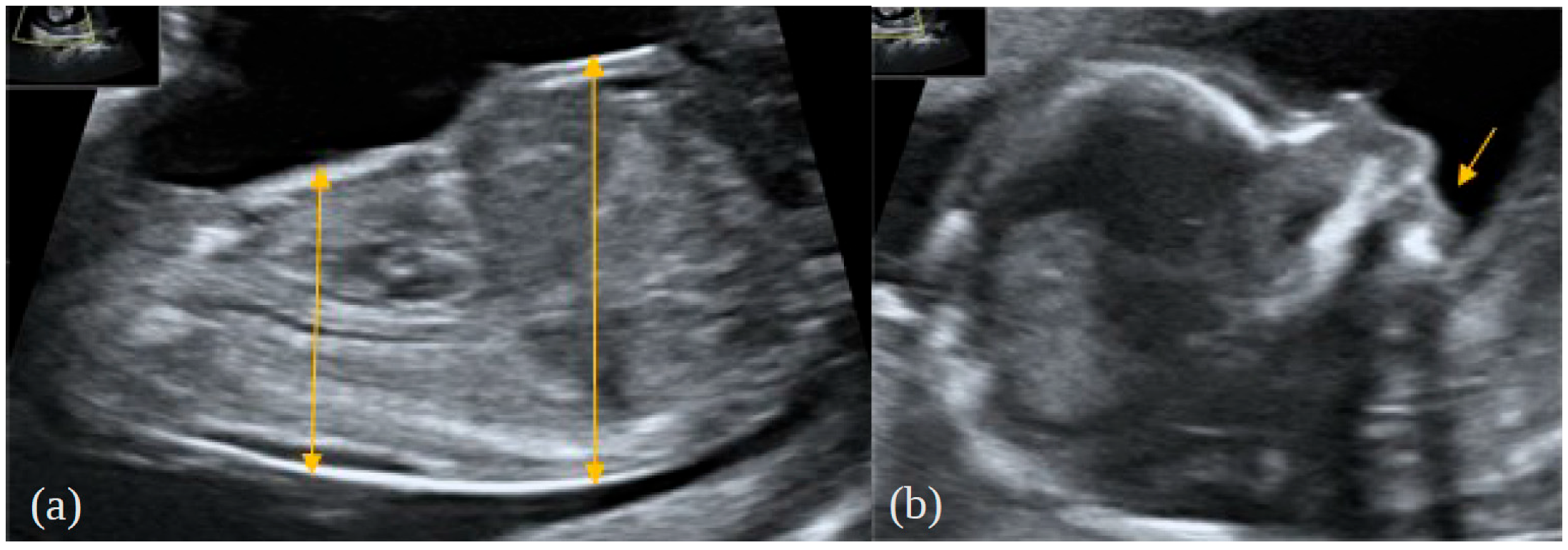
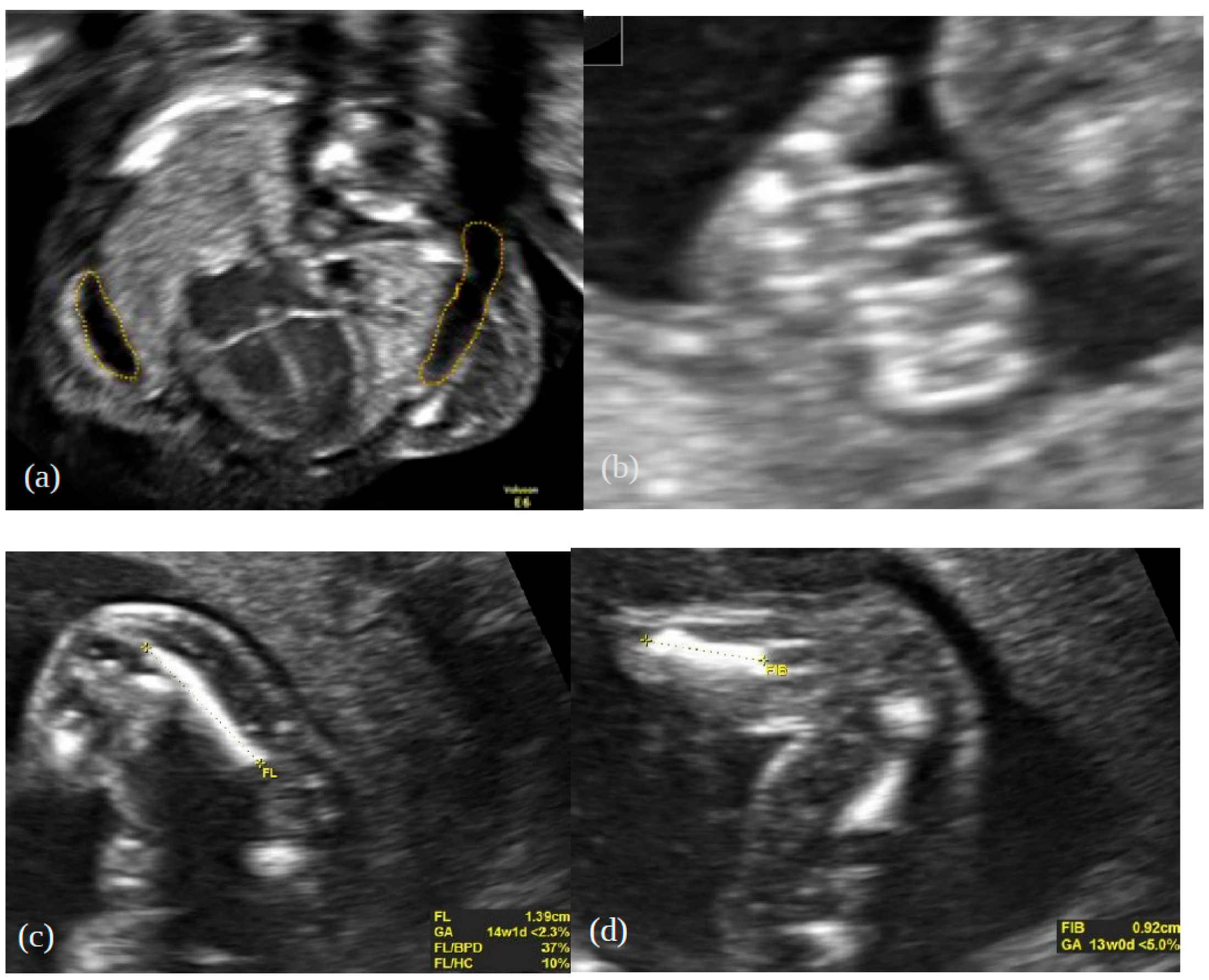
2. Case Report
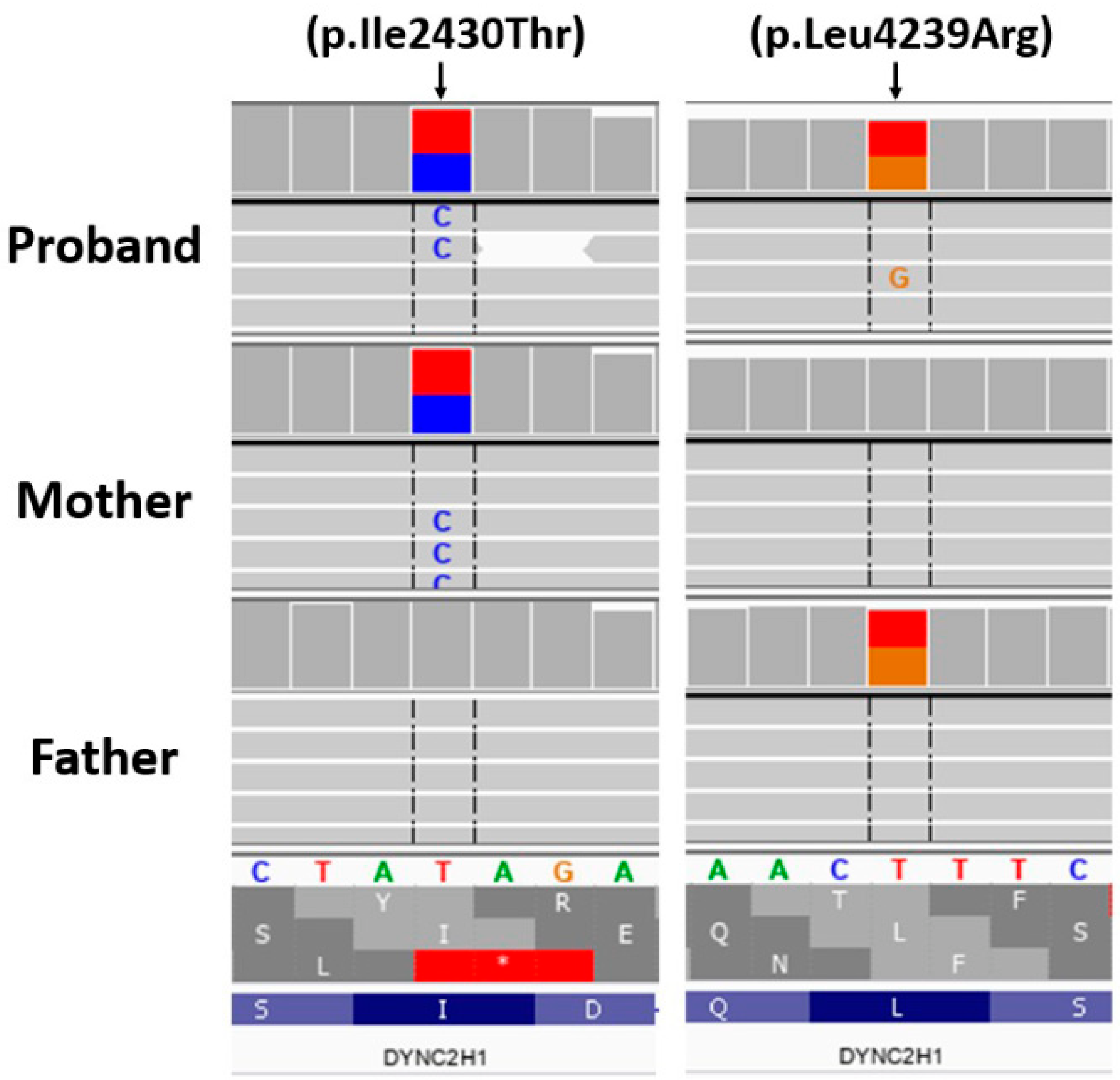
3. Genetic Testing
Materials and Methods
4. Results
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, S.Y.; Jin, D.K. Guidelines for genetic skeletal dysplasias for pediatricians. Ann. Pediatr. Endocrinol. Metab. 2015, 20, 187–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krakow, D.; Rimoin, D.L. The skeletal dysplasias. Genet. Med. 2010, 12, 327–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milks, K.S.; Hill, L.M.; Hosseinzadeh, K. Evaluating skeletal dysplasias on prenatal ultrasound: An emphasis on predicting lethality. Pediatr. Radiol. 2017, 47, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Noel, A.E.; Brown, R.N. Advances in evaluating the fetal skeleton. Int. J. Women’s Health 2014, 13, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Krakow, D.; Lachman, R.S.; Rimoin, D.L. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet. Med. 2009, 11, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Savarirayan, R.; Rossiter, J.P.; Hoover-Fong, J.E.; Irving, M.; Bompadre, V.; Goldberg, M.J.; Bober, M.B.; Cho, T.J.; Kamps, S.E.; Mackenzie, W.G.; et al. Best practice guidelines regarding prenatal evaluation and delivery of patients with skeletal dysplasia. Am. J. Obstet. Gynecol. 2018, 219, 545–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, J.; Yntema, J.L.; van Die, C.E.; Crama, N.; Cornelissen, E.A.; Hamel, B.C. Jeune syndrome: Description of 13 cases and a proposal for follow-up protocol. Eur. J. Pediatr. 2010, 169, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Victoria, T.; Zhu, X.; Lachman, R.; Epelman, M.; Oliver, E.R.; Adzick, N.S.; Biko, D.M. What Is New in Prenatal Skeletal Dysplasias? AJR Am. J. Roentgenol. 2018, 210, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. Part A 2019, 179, 2393–2419. [Google Scholar] [CrossRef] [PubMed]
- Offiah, A.C. Skeletal Dysplasias: An Overview. Endocr. Dev. 2015, 28, 259–276. [Google Scholar] [CrossRef] [PubMed]
- İpek, M.S.; Akgul Ozmen, C. Skeletal Dysplasias That Cause Thoracic Insufficiency in Neonates: Illustrative Case Reports. Medicine 2016, 95, e3298. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, M.; Arts, H.H.; Bongers, E.M.; Yap, Z.; Oud, M.M.; Antony, D.; Duijkers, L.; Emes, R.D.; Stalker, J.; Yntema, J.B.; et al. Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J. Med. Genet. 2013, 50, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Śmigiel, R.; Biela, M.; Szmyd, K.; Błoch, M.; Szmida, E.; Skiba, P.; Walczak, A.; Gasperowicz, P.; Kosińska, J.; Rydzanicz, M.; et al. Rapid Whole-Exome Sequencing as a Diagnostic Tool in a Neonatal/Pediatric Intensive Care Unit. J. Clin. Med. 2020, 9, 2220. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Greenblatt, M.S.; Harrison, S.M.; Nussbaum, R.L.; Prabhu, S.A.; Boucher, K.M.; Biesecker, L.G.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018, 20, 1054–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, K.A.; Suthar, P.P.; Bhesania, S.R.; Patel, A. Antenatal Diagnosis of Jeune Syndrome (Asphyxiating Thoracic Dysplasia) with Micromelia and Facial Dysmorphism on Second-Trimester Ultrasound. Pol. J. Radiol. 2015, 80, 296–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Taylor, S.P.; Ennis, H.A.; Forlenza, K.N.; Duran, I.; Li, B.; Sanchez, J.A.O.; Nevarez, L.; Nickerson, D.A.; Bamshad, M.; et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum. Mutat. 2018, 39, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pan, L.; Teng, Y.; Liang, D.; Li, Z.; Wu, L. Molecular diagnosis for 55 fetuses with skeletal dysplasias by whole-exome sequencing: A retrospective cohort study. Clin. Genet. 2021, 100, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Strong, A.; Li, D.; Mentch, F.; Bedoukian, E.; Hartung, E.A.; Meyers, K.; Skraban, C.; Wen, J.; Medne, L.; Glessner, J.; et al. Ciliopathies: Coloring outside of the lines. Am. J. Med. Genet. Part A 2021, 185, 687–694. [Google Scholar] [CrossRef]
- Phillips, J.D.; van Aalst, J.A. Jeune’s syndrome (asphyxiating thoracic dystrophy): Congenital and acquired. Semin. Pediatr. Surg. 2008, 17, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, H.M.; Valente, E.M. Motile and non-motile cilia in human pathology: From function to phenotypes. J. Pathol. 2017, 241, 294–309, Erratum in J. Pathol. 2017, 241, 564. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [Green Version]
- Dagoneau, N.; Goulet, M.; Geneviève, D.; Sznajer, Y.; Martinovic, J.; Smithson, S.; Huber, C.; Baujat, G.; Flori, E.; Tecco, L.; et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 2009, 84, 706–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badiner, N.; Taylor, S.P.; Forlenza, K.; Lachman, R.S.; University of Washington Center for Mendelian Genomics; Bamshad, M.; Nickerson, D.; Cohn, D.H.; Krakow, D. Mutations in DYNC2H1, the cytoplasmic dynein 2, heavy chain 1 motor protein gene, cause short-rib polydactyly type I, Saldino-Noonan type. Clin. Genet. 2017, 92, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyner, S.E.; Bradshaw, W.T. Jeune syndrome: Considerations for management of asphyxiating thoracic dystrophy. Neonatal. Netw. 2013, 32, 342–352. [Google Scholar] [CrossRef]
- Prasad, P.L.; Prasad, A.N. Jeune Syndrome. Med. J. Armed Forces India 2006, 62, 293–294. [Google Scholar] [CrossRef] [Green Version]
- Saito, W.; Inoue, G.; Imura, T.; Nakazawa, T.; Miyagi, M.; Namba, T.; Shirasawa, E.; Takahira, N.; Takaso, M. Spinal correction of scoliosis in Jeune syndrome: A report of two cases. Scoliosis Spinal Disord. 2016, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Spranger, J.W.; Brill, P.W.; Hall, C.; Nishimura, G.; Superti-Furga, A.; Unger, S. Bone Dysplasias: An Atlas of Genetic Disorders of Skeletal Development, 4th ed.; Oxford University Press: New York, NY, USA, 2018. [Google Scholar] [CrossRef]
- Clark, R.D.; Curry, C.J. Genetic Consultations in the Newborn; Oxford University Press: New York, NY, USA, 2019. [Google Scholar]
- Tüysüz, B.; Bariş, S.; Aksoy, F.; Madazli, R.; Ungür, S.; Sever, L. Clinical variability of asphyxiating thoracic dystrophy (Jeune) syndrome: Evaluation and classification of 13 patients. Am. J. Med. Genet. Part A 2009, 149, 1727–1733. [Google Scholar] [CrossRef]
- Emiralioglu, N.; Wallmeier, J.; Olbrich, H.; Omran, H.; Ozcelik, U. DYNC2H1 mutation causes Jeune syndrome and recurrent lung infections associated with ciliopathy. Clin. Respir. J. 2018, 12, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, M.; Frank, V.; Eisenberger, T.; Al Turki, S.; Bizet, A.A.; Antony, D.; Rix, S.; Decker, C.; Bachmann, N.; Bald, M.; et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney Disease. Hum. Mutat. 2013, 34, 714–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Skeletal Dysplasia | Clinical Description | Inheritance |
|---|---|---|
| Short rib polydactyly ciliopathy syndromes | Constricted thoracic cage, short ribs, short tubular bones, trident appearance of the acetabular roof, polydactyly, and microglossiaSoldino–Noonan and Verma–Naumoff types: marked metaphyseal irregularities; Majewski type: very short, oval tibiae Non-skeletal manifestation: cleft palate, cleft lip, cystic kidneys, genital ambiguity, and foetal hydrops | AR |
| Ellis van Creveld | Short ribs, orodental, cardiac defects, fusion between the hamate and capitate, peculiar deformity of the proximal tibia, short middle phalanges, and postaxial interdigital polydactyly | AR |
| Campomelic dysplasia | Severe limb shortening, tibial bowing, small chest, flat, short vertebrae, hypoplastic scapulae, small iliac wings, and sex reversal | AD |
| Thanatophoric dysplasia type 1 | Severe long bone rhizomelic shortening and sometimes bowing, small chest, frontal forehead prominence, and polyhydramnios Type 1—short, bent femurs like a “French telephone receiver” Type 2—cloverleaf skull | AD |
| Achondroplasia (homozygous) | Limb rhizomelic shortness | AD |
| Achondrogenesis | Short ribs, extremely short limbs, flat face, hydrops, decreased ossification of skull and vertebral bodies, and ossified pubic bones | AD |
| Atelosteogenesis | Severe shortening of limbs, hypoplasia of the humeri, femurs, thoracic spine, dislocated elbows, hips, knee, and possible lack of ossification of single hand bones | AD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stembalska, A.; Rydzanicz, M.; Klaniewska, M.; Dudarewicz, L.; Pollak, A.; Biela, M.; Stawinski, P.; Ploski, R.; Smigiel, R. Prenatal Diagnosis of Jeune Syndrome Caused by Compound Heterozygous Variants in DYNC2H1 Gene—Case Report with Rapid WES Procedure and Differential Diagnosis of Lethal Skeletal Dysplasias. Genes 2022, 13, 1339. https://doi.org/10.3390/genes13081339
Stembalska A, Rydzanicz M, Klaniewska M, Dudarewicz L, Pollak A, Biela M, Stawinski P, Ploski R, Smigiel R. Prenatal Diagnosis of Jeune Syndrome Caused by Compound Heterozygous Variants in DYNC2H1 Gene—Case Report with Rapid WES Procedure and Differential Diagnosis of Lethal Skeletal Dysplasias. Genes. 2022; 13(8):1339. https://doi.org/10.3390/genes13081339
Chicago/Turabian StyleStembalska, Agnieszka, Małgorzata Rydzanicz, Magdalena Klaniewska, Lech Dudarewicz, Agnieszka Pollak, Mateusz Biela, Piotr Stawinski, Rafal Ploski, and Robert Smigiel. 2022. "Prenatal Diagnosis of Jeune Syndrome Caused by Compound Heterozygous Variants in DYNC2H1 Gene—Case Report with Rapid WES Procedure and Differential Diagnosis of Lethal Skeletal Dysplasias" Genes 13, no. 8: 1339. https://doi.org/10.3390/genes13081339