
McCune-Albright Syndrome in Infant with Growth Hormone Excess

 , ,
, ,
Abstract
:1. Introduction
2. Results
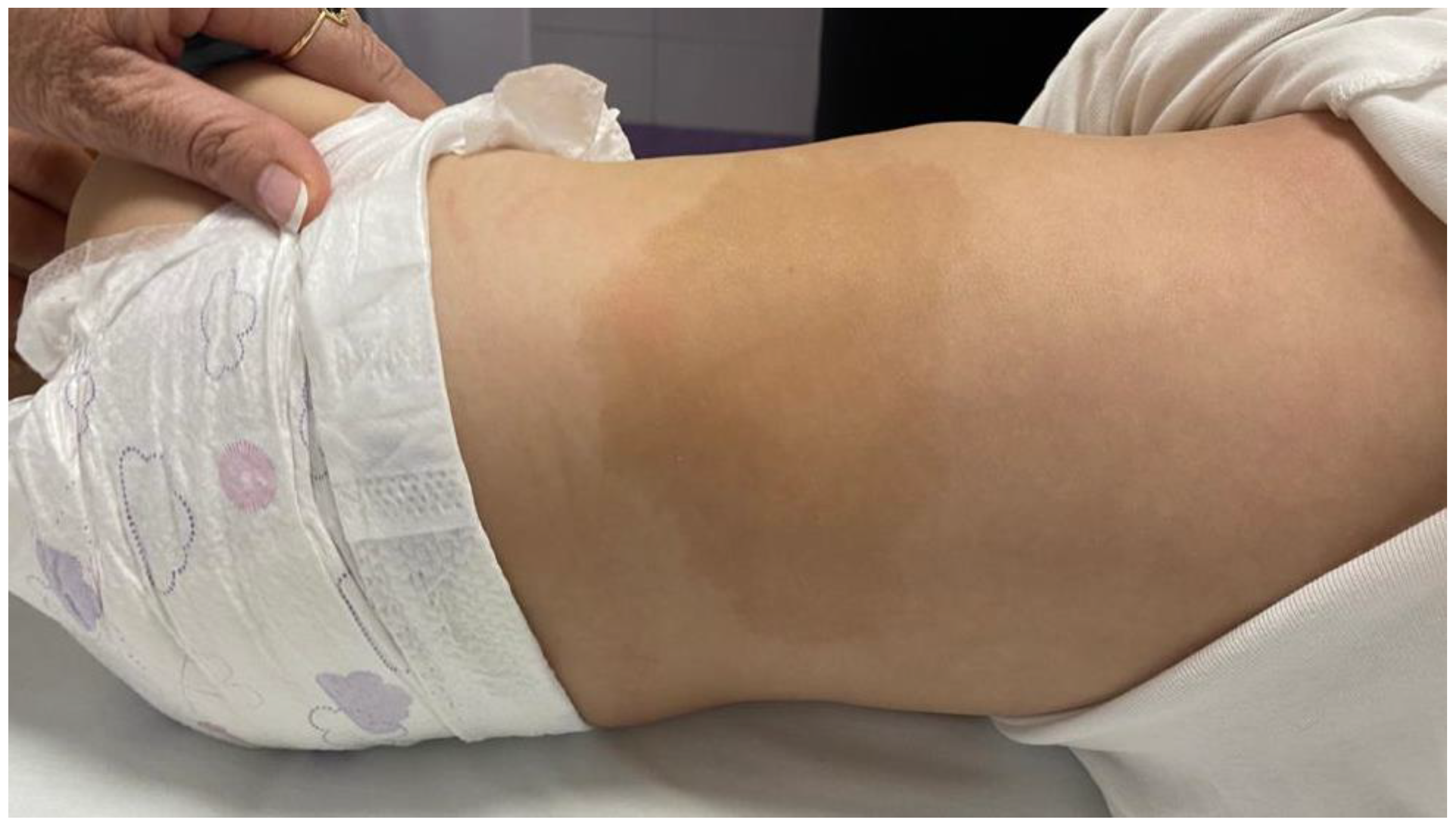
2.1. Anamnestic Data and Clinical Examination
2.2. Assessment of Hormonal Status
2.3. Diagnostic Imaging
2.4. Genetic Analysis
2.5. Treatment
2.6. Hormonal Status Follow-Up
2.7. Growth Rate Follow-Up
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spencer, T.; Pan, K.S.; Collins, M.T.; Boyce, A.M. The Clinical Spectrum of McCune-Albright Syndrome and Its Management. Horm. Res. Paediatr. 2019, 92, 347–356. [Google Scholar] [CrossRef]
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright syndrome. Orphanet J. Rare Dis. 2008, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Robinson, C.; Collins, M.T.; Boyce, A.M. Fibrous dysplasia/McCune-Albright syndrome: Clinical and translational perspectives. Curr. Osteoporos. Rep. 2016, 14, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef]
- Hayward, B.E.; Kamiya, M.; Strain, L.; Moran, V.; Campbell, R.; Hayashizaki, Y.; Bonthron, D.T. The human GNAS1 gene is imprinted and encodes distinct paternally and biallelically expressed G proteins. Proc. Natl. Acad. Sci. USA 1998, 18, 10038–10043. [Google Scholar] [CrossRef] [Green Version]
- Diaz, A.; Danon, M.; Crawford, J. McCune-Albright Syndrome and Disorders Due to Activating Mutations of GNAS1. J. Pediatr. Endocrinol. Metab. 2007, 20, 853–880. [Google Scholar] [CrossRef]
- Christoforidis, A.; Maniadaki, I.; Stanhope, R. McCune-Albright syndrome: Growth hormone and prolactin hypersecretion. J. Pediatr. Endocrinol. Metab. 2006, 19 (Suppl. 2), 623–625. [Google Scholar] [CrossRef]
- Akintoye, S.O.; Chebli, C.; Booher, S.; Feuillan, P.; Kushner, H.; Leroith, D.; Cherman, N.; Bianco, P.; Wientroub, S.; Robey, P.G.; et al. Characterization of gsp-mediated growth hormone excess in the context of McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 2002, 87, 5104–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumbroso, S.; Paris, F.; Sultan, C. European Collaborative Study. Activating Gsalpha mutations: Analysis of 113 patients with signs of McCune-Albright syndrome-a European Collaborative Study. J. Clin. Endocrinol. Metab. 2004, 89, 2107–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corica, D.; Aversa, T.; Pepe, G.; De Luca, F.; Wasniewska, M. Peculiarities of Precocious Puberty in Boys and Girls With McCune-Albright Syndrome. Front. Endocrinol. 2018, 9, 337. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.J.; Kelly, M.H.; Collins, M.T. Cushing syndrome in the McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Mastorakos, G.; Mitsiades, N.S.; Doufas, A.G.; Koutras, D.A. Hyperthyroidism in McCune-Albright syndrome with a review of thyroid abnormalities sixty years after the first report. Thyroid Off. J. Am. Thyroid Assoc. 1997, 7, 433–439. [Google Scholar] [CrossRef]
- Tetlow, L.J.; Clayton, P.E. Tests and normal values in pediatric endocrinology. In Brook’s Clinical Pediatric Endocrinology, 5th ed.; Brook, C., Clayton, P., Brown, R., Eds.; Wiley-Blackwell Publication: Hoboken, NJ, USA, 2008; pp. 530–531. [Google Scholar]
- Collins, M.T.; Sarlis, N.J.; Merino, M.J.; Monroe, J.; Crawford, S.E.; Krakoff, J.A.; Guthrie, L.C.; Bonat, S.; Robey, P.G.; Shenker, A. Thyroid Carcinoma in the McCune-Albright Syndrome: Contributory Role of Activating Gsα Mutations. J. Clin. Endocrinol. Metab. 2003, 88, 4413–4417. [Google Scholar] [CrossRef] [Green Version]
- Huston, T.L.; Simmons, R.M. Ductal carcinoma in situ in a 27-year-old woman with McCune-Albright syndrome. Breast J. 2004, 10, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Hagelstein-Rotman, M.; Meier, M.E.; Majoor, B.C.J.; Cleven, A.H.G.; Dijkstra, P.D.S.; Hamdy, N.A.T.; van de Sande, M.A.J.; Dekkers, O.M.; Appelman-Dijkstra, N.M. Increased Prevalence of Malignancies in Fibrous Dysplasia/McCune-Albright Syndrome (FD/MAS): Data from a National Referral Center and the Dutch National Pathology Registry (PALGA). Calcif. Tissue Int. 2021, 108, 346–353. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, Y.; Wang, L.; Deng, K.; Yang, H.; Lu, L.; Feng, F.; Xing, B.; You, H.; Jin, Z.; et al. Clinical characteristics and management of growth hormone excess in patients with McCune-Albright syndrome. Eur. J. Endocrinol. 2017, 176, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Salenave, S.; Boyce, A.M.; Collins, M.T.; Chanson, P. Acromegaly and McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 2014, 99, 1955–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, X.; Duan, L.; Yao, Y.; Xing, B.; Deng, K.; Wang, L.; Feng, F.; Liang, Z.; You, H.; Yang, H.; et al. Clinical characteristics and management of patients with McCune-Albright syndrome with GH excess and precocious puberty: A case series and literature review. Front. Endocrinol. 2021, 12, 672394. [Google Scholar] [CrossRef] [PubMed]
- Tessaris, D.; Boyce, A.M.; Zacharin, M.; Matarazzo, P.; Lala, R.; De Sanctis, L.; Collins, M.T. Growth hormone-Insulin-like growth factor 1 axis hyperactivity on bone fibrous dysplasia in McCune-Albright Syndrome. Clin. Endocrinol 2018, 89, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. Insulin-like growth factor 1 (IGF-1): A growth hormone. Mol. Pathol. 2001, 54, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Al-Samerria, S.; Radovick, S. The role of insulin-like growth factor-1 (IGF-1) in the control of neuroendocrine regulation of growth. Cells 2021, 10, 2664. [Google Scholar] [CrossRef] [PubMed]
- Racine, H.L.; Serrat, M.A. The actions of IGF-1 in the growth plate and its role in postnatal bone elongation. Curr. Osteoporos. Rep. 2020, 18, 210–227. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.; Cheng, C.M.; Kopchick, J.J.; Bondy, C.A. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J. Endocrinol. 2004, 180, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Tahimic, C.G.T.; Wang, Y.; Bikle, D.D. Anabolic effects of IGF-1 signaling on the skeleton. Front. Endocrinol. 2013, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Yang, W.; De Luca, F. Insulin-like growth factor-independent effects of growth hormone on growth plate chondrogenesis and longitudinal bone growth. Endocrinology 2015, 156, 2541–2551. [Google Scholar] [CrossRef] [Green Version]
- Dobie, R.; Ahmed, S.F.; Staines, K.A.; Pass, C.; Jasim, S.; MacRae, V.E.; Farquharson, C. Increased linear bone growth by GH in the absence of SOCS2 is independent of IGF-1: SOCS2 REGULATION OF GH INDUCED GROWTH. J. Cell Physiol. 2015, 230, 2796–2806. [Google Scholar] [CrossRef] [Green Version]
- Zacharin, M. Paediatric management of endocrine complications in McCune-Albright syndrome. J. Pediatr. Endocrinol. Metab. 2005, 18, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Nozières, C.; Berlier, P.; Dupuis, C.; Raynaud-Ravni, C.; Morel, Y.; Chazot, F.B.; Nicolino, M. Sporadic and genetic forms of paediatric somatotropinoma: A retrospective analysis of seven cases and a review of the literature. Orphanet J. Rare Dis. 2011, 6, 67. [Google Scholar] [CrossRef] [Green Version]
- Akintoye, S.O.; Kelly, M.H.; Brillante, B.; Cherman, N.; Turner, S.; Butman, J.A.; Robey, P.G.; Collins, M.T. Pegvisomant for the treatment of gsp-mediated growth hormone excess in patients with McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 2960–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Main, K.M.; Sehested, A.; Feldt-Rasmussen, U. Pegvisomant treatment in a 4-year-old girl with neurofibromatosis type 1. Horm. Res. 2006, 65, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, N.; Racine, M.S.; Thomas, P.; Degnan, B.; Chandler, W.; Barkan, A. Treatment of pituitary gigantism with the growth hormone receptor antagonist pegvisomant. J. Clin. Endocrinol. Metab. 2008, 93, 2953–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergamaschi, S.; Ronchi, C.L.; Giavoli, C.; Ferrante, E.; Verrua, E.; Ferrari, D.I.; Lania, A.; Rusconi, R.; Spada, A.; Beck-Peccoz, P. Eight-year follow-up of a child with a GH/prolactin-secreting adenoma: Efficacy of pegvisomant therapy. Horm. Res. Paediatr. 2010, 73, 74–79. [Google Scholar] [CrossRef]
- Kovacs, K.; Horvath, E.; Thorner, M.O.; Rogol, A.D. Mammosomatotroph hyperplasia associated with acromegaly and hyperprolactinemia in a patient with the McCune-Albright syndrome: A histologic, immunocytologic and ultrastructural study of the surgically-removed adenohypophysis. Vichows. Arch. A Pathol. Anat. 1984, 403, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.M.W.; Brain, C.E.; Carson, D.J.; Hyde, J.C.; Grant, D.B. Cushing’s syndrome caused by nodular adrenal hyperplasia in children with McCune-Albright syndrome. J. Pediatr. 1999, 134, 789–792. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Age at Measurement | 8.9 Months | 9.1 Months | 1.39 Years | 2 Years | Normal Range |
|---|---|---|---|---|---|
| IGF-1 (nmol/L) | 8.7 (ref. 2.34–18.98) | 8.32 (ref. 2.34–18.98) | 5.52 (ref. 2.6–20.67) | 5.02 (ref. 2.6–20.67) | depending on age |
| IGFBP-3 (nmol/L) | 2.73 | / | / | / | depending on age |
| FSH (IU/L) | 5.89 | / | 8.04 | 10.6 | 0.2–11.1 |
| LH (IU/L) | <0.3 | / | <0.3 | 0.312 | 0.312 |
| E2 (pmol/L) | <18.4 | / | <18.4 | <18.4 | <37.7 |
| PRL (mIU/L) | 300 | 903 | 427 | 303 | 102–496 |
| ACTH (pmol/L) | 11.6 | / | 22.8 | 6.5 | 1.6–13.9 |
| cortisol (nmol/L) | 288 | / | 517 | 224 | 171–536 |
| TSH (mIU/L) | / | / | 1.36 | 1.87 | 0.7–5.97 |
| T4 (nmol/L) | / | / | 80.6 | 108 | 76.6–189 |
| PTH (pmol/L) | 3.4 | / | 4.2 | / | 1.58–6.03 |
| Time (Minutes) | 0 | 30 | 60 | 90 | 120 |
|---|---|---|---|---|---|
| Glucose (mmol/L) | 4.23 | 5.55 | 4.73 | 5.25 | 3.64 |
| Insulin (uU/mL) | 2.4 | 10.8 | 5.9 | 8.8 | 1.2 |
| Growth hormone (mU/L) | 6.45 | 2.82 | 3.64 | 3.33 | 10.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzica, K.; Simunovic, M.; Ivancic, M.; Tudor, D.; Skrabic, I.; Skrabic, V. McCune-Albright Syndrome in Infant with Growth Hormone Excess. Genes 2022, 13, 1345. https://doi.org/10.3390/genes13081345
Brzica K, Simunovic M, Ivancic M, Tudor D, Skrabic I, Skrabic V. McCune-Albright Syndrome in Infant with Growth Hormone Excess. Genes. 2022; 13(8):1345. https://doi.org/10.3390/genes13081345
Chicago/Turabian StyleBrzica, Katarina, Marko Simunovic, Matea Ivancic, Darija Tudor, Ivna Skrabic, and Veselin Skrabic. 2022. "McCune-Albright Syndrome in Infant with Growth Hormone Excess" Genes 13, no. 8: 1345. https://doi.org/10.3390/genes13081345
APA StyleBrzica, K., Simunovic, M., Ivancic, M., Tudor, D., Skrabic, I., & Skrabic, V. (2022). McCune-Albright Syndrome in Infant with Growth Hormone Excess. Genes, 13(8), 1345. https://doi.org/10.3390/genes13081345