miR-23b-3p Modulating Cytoprotective Autophagy and Glutamine Addiction in Sorafenib Resistant HepG2, a Hepatocellular Carcinoma Cell Line


Abstract
:
1. Introduction
2. Materials and Methodology
2.1. Chemicals
2.2. Cell Lines
- Fold resistance = IC-50 value of resistant cells/IC-50 value of parental cells
2.3. RNA Isolation and Its Quantification, cDNA Synthesis, and qRT-PCR
2.4. MTT Assay
- % Cell viability = ((Absorbance of test − Absorbance of Blank)/(Absorbance of control − Absorbance of Blank)) * 100
2.5. Annexin V/Propidium Iodide (PI)
2.6. Green Autophagy Dye Assay
2.7. Next Generation Sequencing (NGS)
2.8. In Silico Analysis
2.9. Transfection of miRNA Mimics and Antimirs
2.10. MiRNA Isolation; Polyadenylation, cDNA Synthesis Poly A Tailed miRNA, qRT-PCR for miRNAs
2.11. Statistical Analysis
3. Results
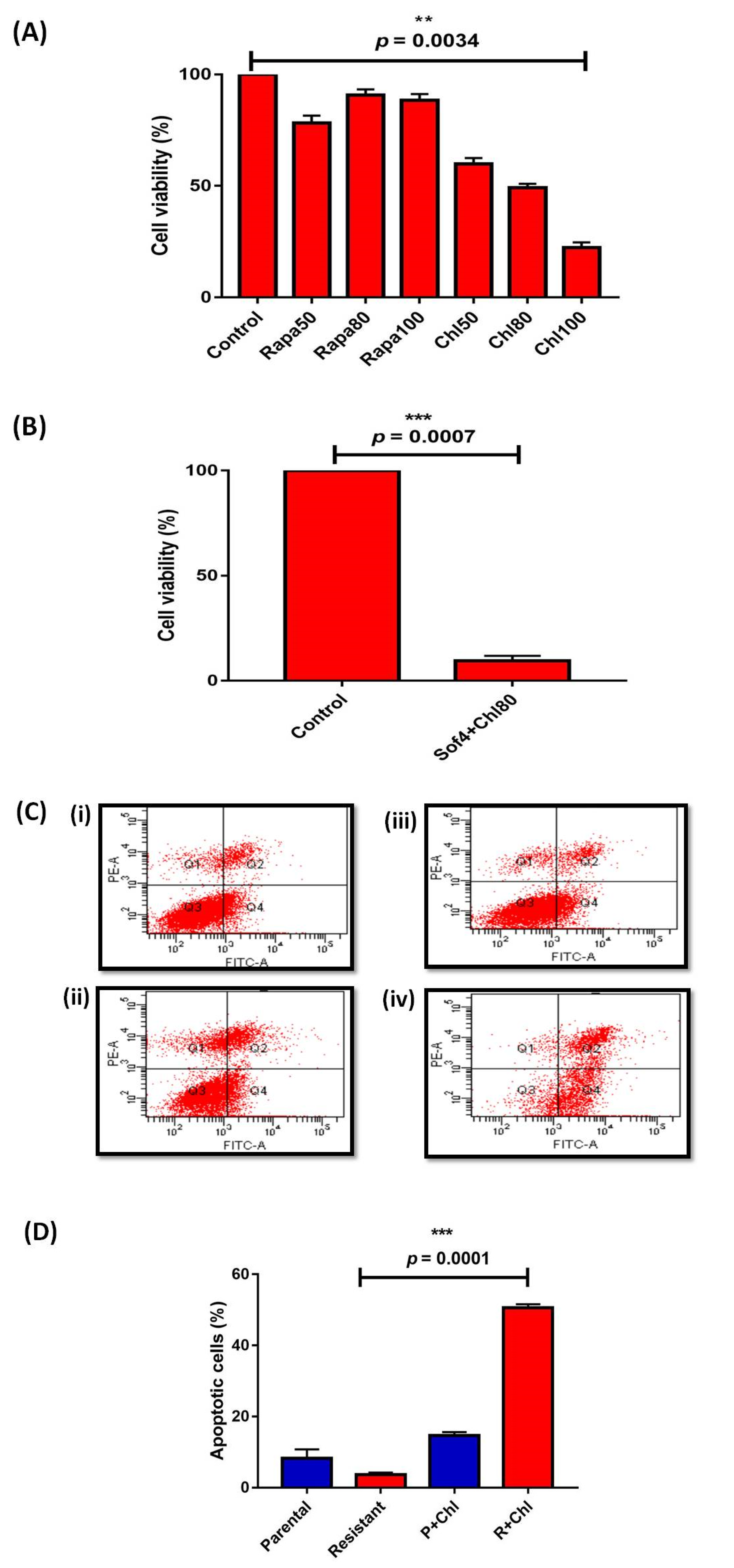
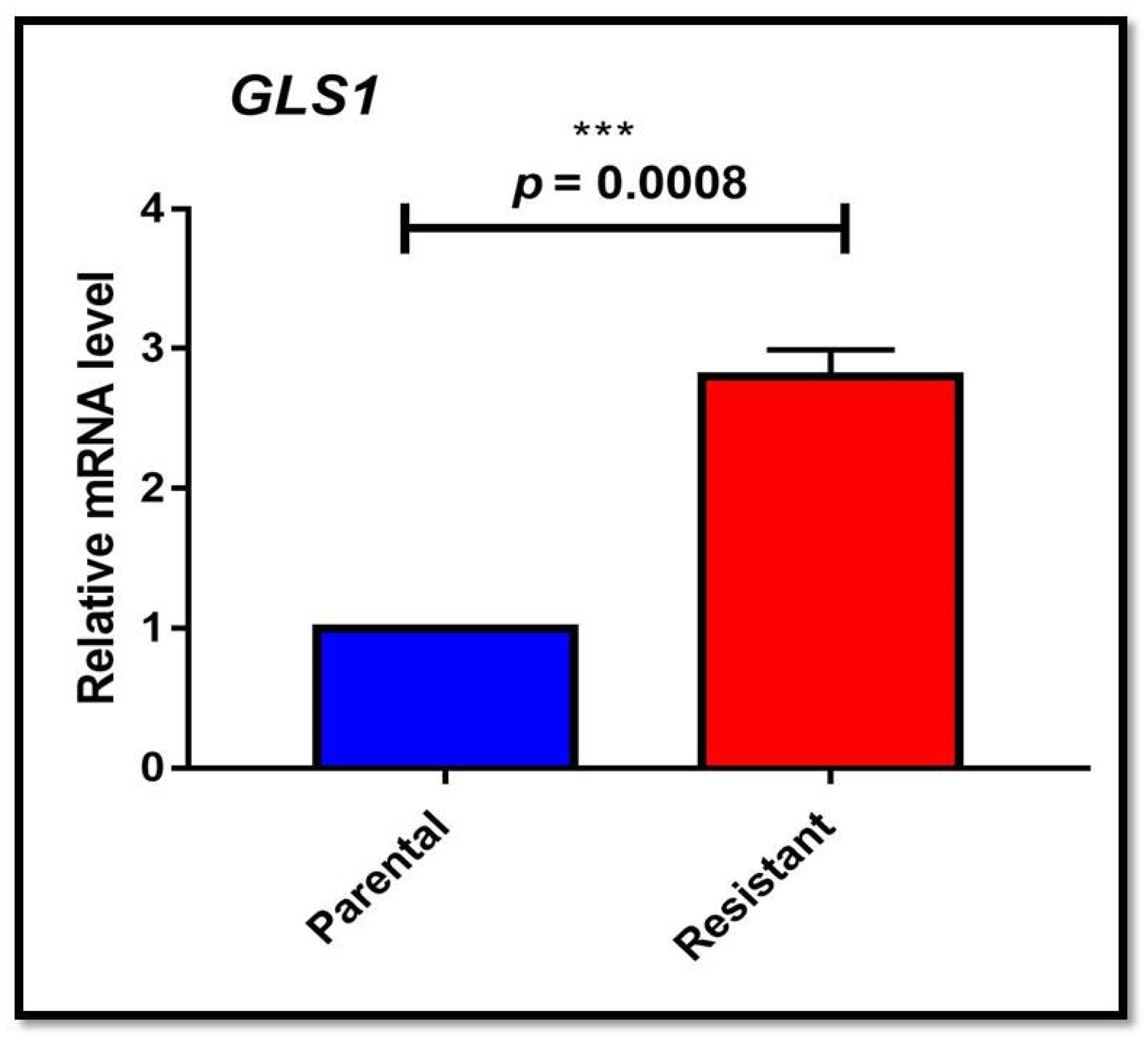
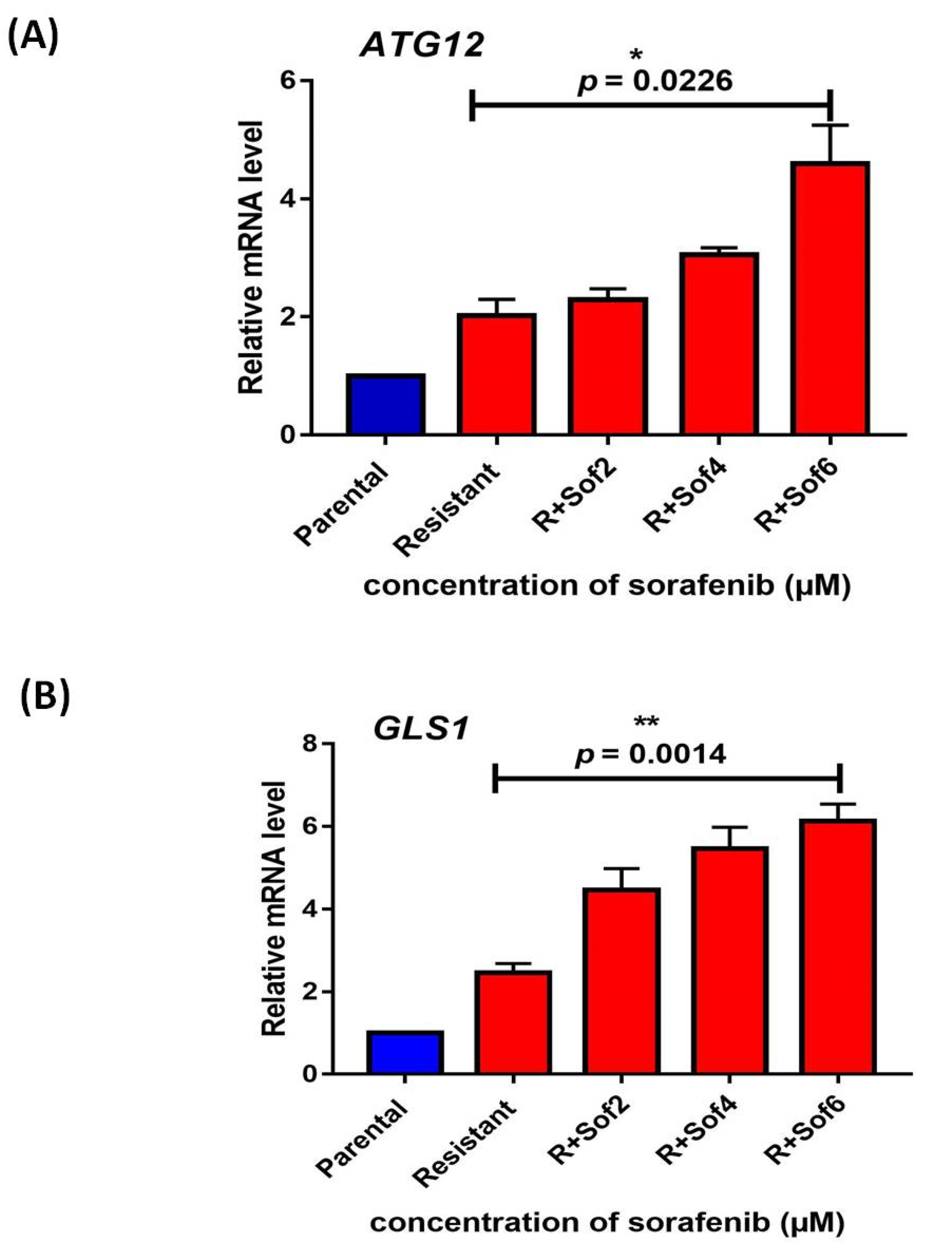
3.1. Autophagy and Glutamine Addiction as Cytoprotective in Sorafenib-Resistant HepG2 Cells
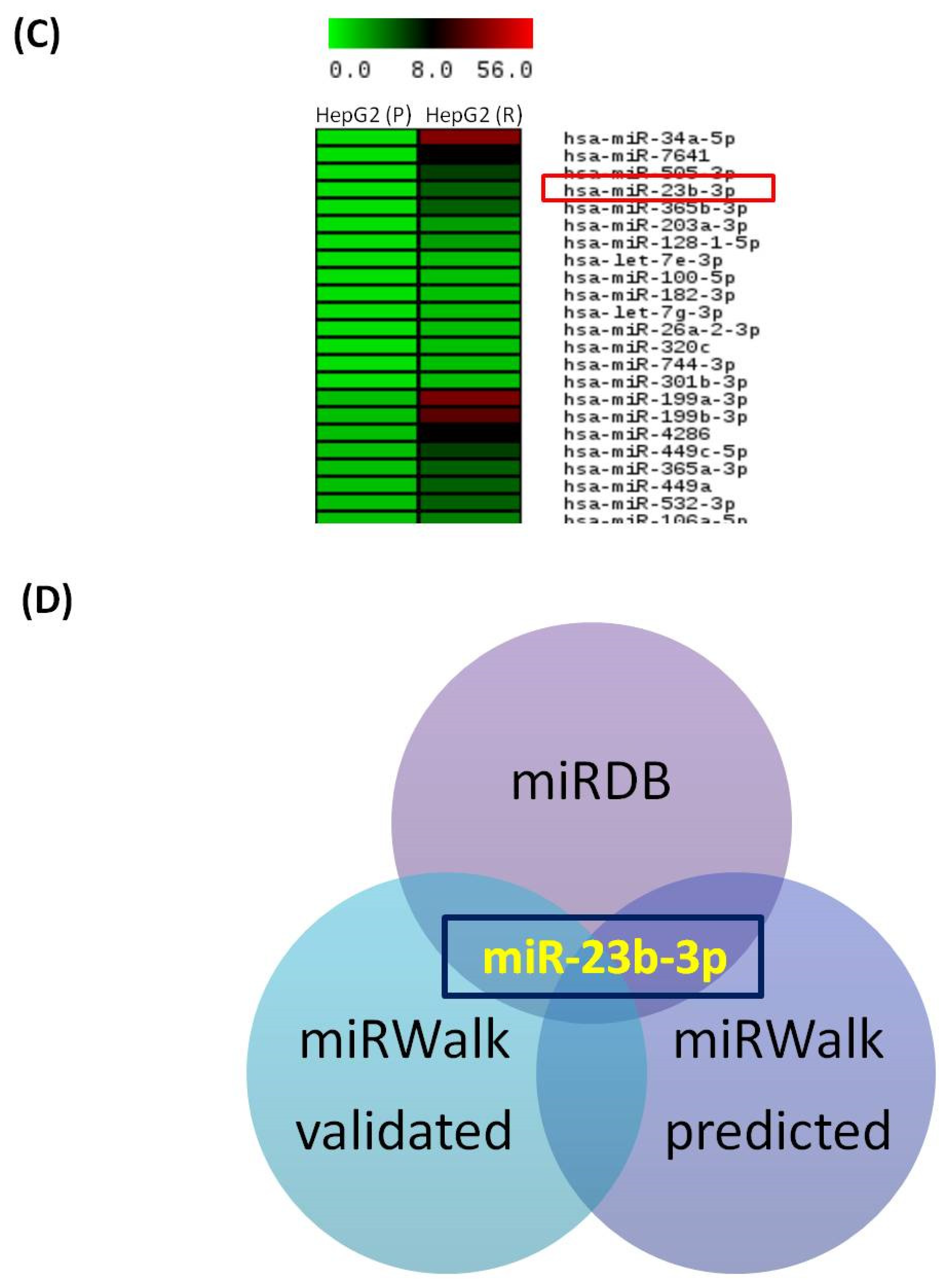
3.2. Autophagy and Glutamine Addiction-Specific microRNAs and their Target Genes in Sorafenib-Resistant HCC Cell Lines
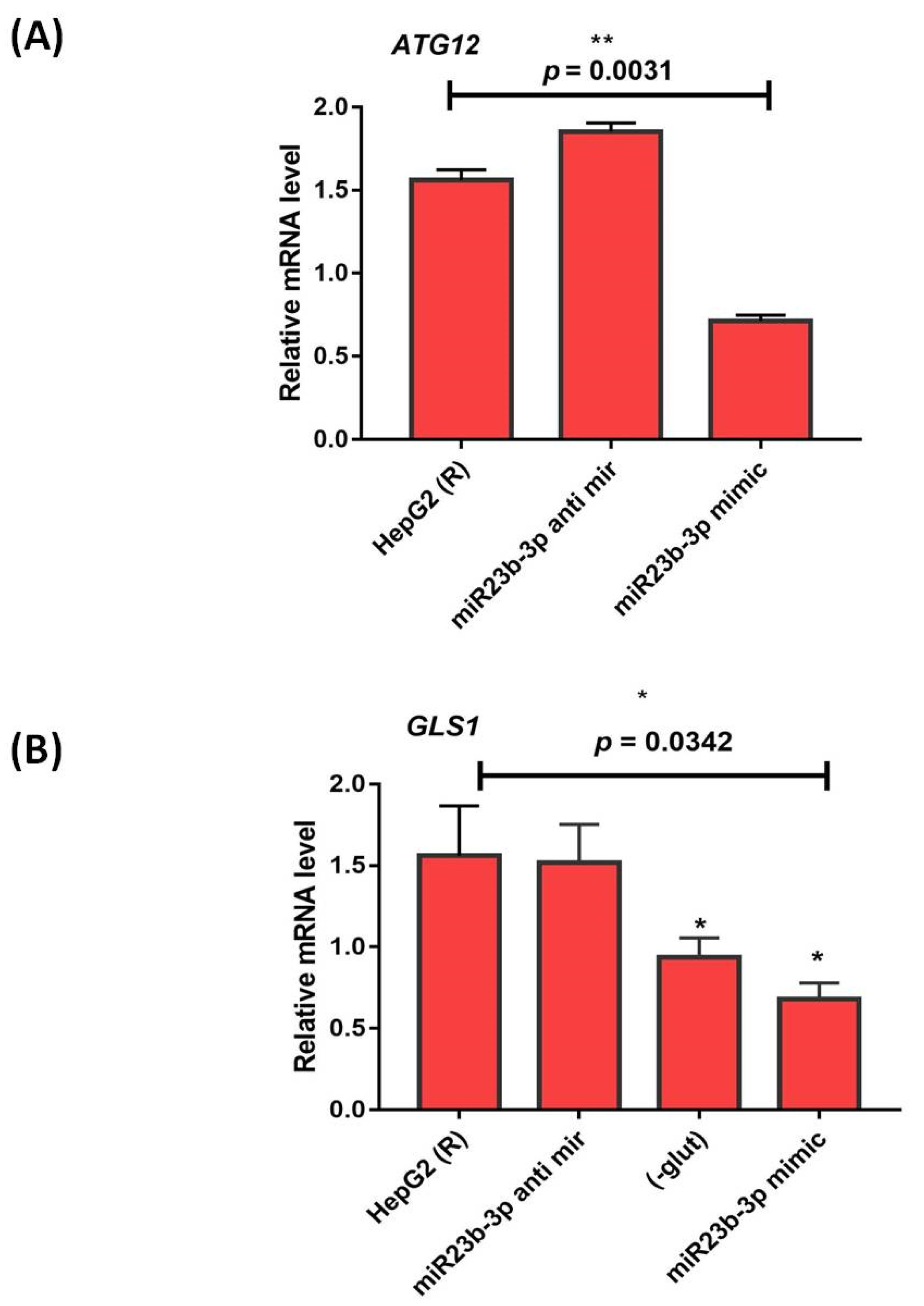
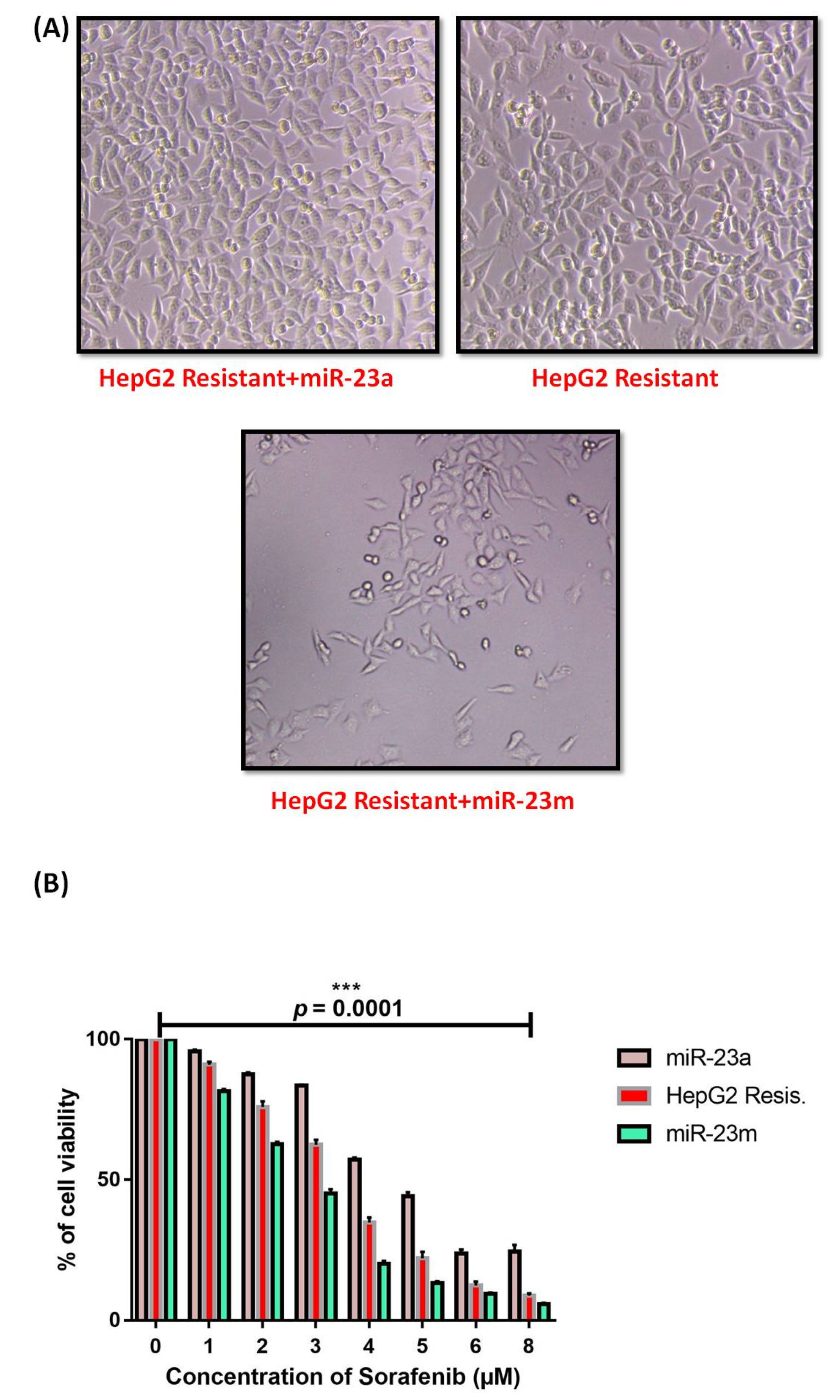
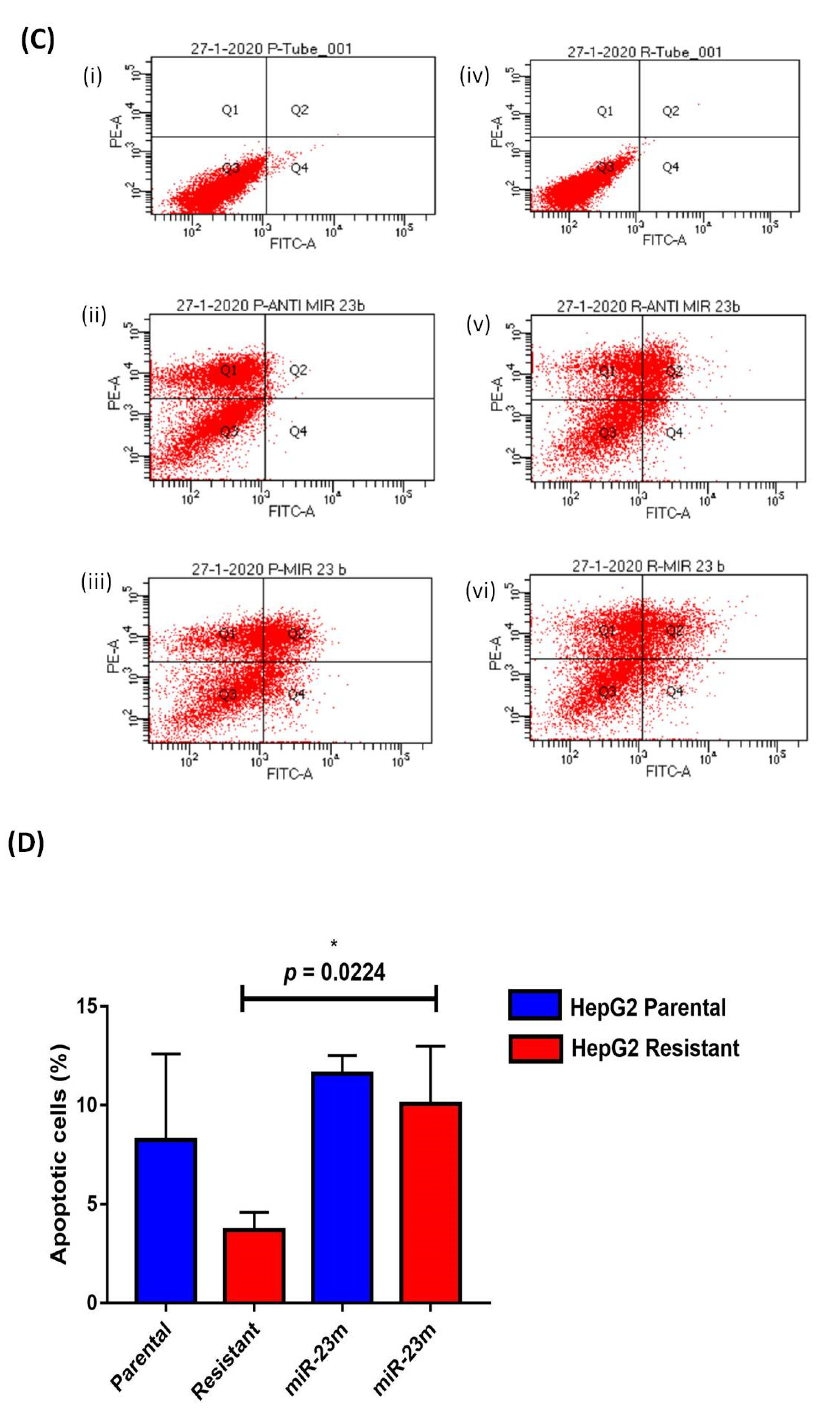
3.3. Functional Analysis of miR-23b-3p
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertuccio, P.; Turati, F.; Carioli, G.; Rodriguez, T.; La Vecchia, C.; Malvezzi, M.; Negri, E. Global trends and predictions in hepatocellular carcinoma mortality. J. Hepatol. 2017, 67, 302–309. [Google Scholar] [CrossRef]
- Acharya, S.K. Epidemiology of hepatocellular carcinoma in India. J. Clin. Exp. Hepatol. 2014, 4 (Suppl. 3), S27–S33. [Google Scholar] [CrossRef] [Green Version]
- Sarin, S.K.; Thakur, V.; Guptan, R.C.; Saigal, S.; Malhotra, V.; Thyagarajan, S.P.; Das, B.C. Profile of hepatocellular carcinoma in India: An insight into the possible etiologic associations. J. Gastroenterol. Hepatol. 2001, 16, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar] [PubMed]
- O’Connor, R.; Clynes, M.; Dowling, P.; O’Donovan, N.; O’Driscoll, L. Drug resistance in cancer—Searching for mechanisms, markers and therapeutic agents. Expert. Opin. Drug Metab. Toxicol. 2007, 3, 805–817. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [Green Version]
- Makol, A.; Kaur, H.; Sharma, S.; Kanthaje, S.; Kaur, R.; Chakraborti, A. Vimentin as a potential therapeutic target in sorafenib resistant HepG2, a HCC model cell line. Clin. Mol. Hepatol. 2020, 26, 45–53. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Pan, Y.; Liang, Y.; Xu, J.; Cai, X. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2020, 51, 102610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, W.-T.; Shiau, C.-W.; Chen, H.-L.; Liu, C.-Y.; Lin, C.-S.; Cheng, A.-L.; Chen, P.-J.; Chen, K.-F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Q.; Song, J. Inhibition of autophagy potentiates the proliferation inhibition activity of microRNA-7 in human hepatocellular carcinoma cells. Oncol. Lett. 2017, 14, 3566–3572. [Google Scholar] [CrossRef] [Green Version]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Yin, D.; Wang, L.; Liu, H.; Tian, L.; Fang, X.; Meng, X.; et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1α inhibition in hepatocellular carcinoma. Hepatology 2013, 57, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 1955, 122, 501–514. [Google Scholar] [CrossRef]
- Yu, D.; Shi, X.; Meng, G.; Chen, J.; Yan, C.; Jiang, Y.; Wei, J.; Ding, Y. Kidney-type glutaminase (GLS1) is a biomarker for pathologic diagnosis and prognosis of hepatocellular carcinoma. Oncotarget 2015, 6, 7619–7631. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulphide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanthaje, S.; Makol, A.; Chakraborti, A. Sorafenib response in hepatocellular carcinoma: MicroRNAs as tuning forks. Hepatol. Res. 2018, 48, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin, V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar]
- Ma, J.; Mannoor, K.; Gao, L.; Tan, A.; Guarnera, M.A.; Zhan, M.; Shetty, A.; Stass, S.A.; Xing, L.; Jiang, F. Characterization of microRNA transcriptome in lung cancer by next-generation deep sequencing. Mol. Oncol. 2014, 8, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical Aspects of microRNA Target Prediction. Curr. Mol. Med. 2011, 11, 93–109. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; Oliveira, A.C.d.; Santoro, A.; Raoul, J.-L.; Forner, A. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Chaudhuri, T.; Lokesh, K.; Lakshmaiah, K.; Babu, K.G.; Dasappa, L.; Jacob, L.A.; Babu, M.S.; Rudresha, A.; Rajeev, L. Advanced hepatocellular carcinoma: A regional cancer center experience of 48 cases. Indian J. Cancer 2017, 54, 526–529. [Google Scholar] [CrossRef]
- Manov, I.; Pollak, Y.; Broneshter, R.; Iancu, T.C. Inhibition of doxorubicin-induced autophagy in hepatocellular carcinoma Hep3B cells by sorafenib--the role of extracellular signal-regulated kinase counteraction. FEBS J. 2011, 278, 3494–3507. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Wu, L.; Pei, M.; Li, H.; Jiang, Y. miR-145 inhibits glutamine metabolism through c-myc/GLS1 pathways in ovarian cancer cells. Cell Biol. Int. 2019, 43, 921–930. [Google Scholar] [CrossRef]
- Zhu, Y.; Lin, G.; Dai, Z.; Zhou, T.; Li, T.; Yuan, T.; Wu, Z.; Wu, G.; Wang, J. L-Glutamine deprivation induces autophagy and alters the mTOR and MAPK signaling pathways in porcine intestinal epithelial cells. Amino Acids 2015, 47, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Ye, X.; Ma, X.; Hu, X.; Yang, W.; Shi, J.; Chen, G.; Gong, L. SNGH16 regulates cell autophagy to promote Sorafenib Resistance through suppressing miR-23b-3p via sponging EGR1 in hepatocellular carcinoma. Cancer Med. 2020, 9, 4324–4338. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Zhang, Z.; Shang, Y.; Jiang, X.; Dong, J.; Yu, P.; Nie, Y.; Zhao, Q. miR-23b-3p regulates the chemoresistance of gastric cancer cells by targeting ATG12 and HMGB2. Cell Death Dis. 2015, 6, e1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, R.; Li, X.; Ma, Y. miR-23b-3p suppressing PGC1α promotes proliferation through reprogramming metabolism in osteosarcoma. Cell Death Dis. 2019, 10, 381. [Google Scholar] [CrossRef] [PubMed]
- He, R.Q.; Wu, P.R.; Xiang, X.L.; Yang, X.; Liang, H.-W.; Qiu, X.-H.; Yang, L.-H.; Peng, Z.-G.; Chen, G. Downregulated miR-23b-3p expression acts as a predictor of hepatocellular carcinoma progression: A study based on public data and RT-qPCR verification. Int. J. Mol. Med. 2018, 41, 2813–2831. [Google Scholar] [CrossRef] [Green Version]
- Manganelli, M.; Grossi, I.; Ferracin, M.; Guerriero, P.; Negrini, M.; Ghidini, M.; Senti, C.; Ratti, M.; Pizzo, C.; Passalacqua, R.; et al. Longitudinal Circulating Levels of miR-23b-3p, miR-126-3p and lncRNA GAS5 in HCC Patients Treated with Sorafenib. Biomedicines 2021, 13, 813. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Product Size (bp) | Thermal Conditions |
|---|---|---|---|
| ATG7 | FP: ATGGTGCTGGTTTCCTTGCT RP: CTGCTACTCCATCTGTGGGC | 158 bp | 95 °C-10 s; 54 °C-15 s; 72 °C-20 s |
| ATG12 | FP: GCGAACACGAACCATCCAAG RP: CACGCCTGAGACTTGCAGTA | 189 bp | 95 °C-10 s; 54 °C-15 s; 72 °C-20 s |
| Beclin1 | FP: ACCGTGTCACCATCCAGGAA RP: GAAGCTGTTGGCACTTTCTGT | 231 bp | 95 °C-10 s; 54 °C-15 s; 72 °C-20 s |
| LC3II | FP: GATGTCCGACTTATTCGAGAGC RP: TTGAGCTGTAAGCGCCTTCTA | 167 bp | 95 °C-10 s; 59 °C-15 s; 72 °C-20 s |
| c-MYC | FP: TAGTGGAAAACCAGCCTCCC RP: CCGAGTCGTAGTCGAGGTCA | 81 bp | 95 °C-10 s; 56 °C-15 s; 72 °C-20 s |
| GLS1 | FP: TCCCCAAGGACAGGTGGAA RP: ACGGTTTGATTTTCCTTCCCG | 142 bp | 95 °C-10 s; 64 °C-15 s; 72 °C-20 s |
| GAPDH | FP: CCATCTTCCAGGAGCGAGA RP: GGTCATGAGTCCTTCCACGAT | 305 bp | 95 °C-10 s; 60 °C-15 s; 72 °C-20 s |
| miRNA | Primer Sequence |
|---|---|
| hsa-miR-23b-3p | FP-ATCACATTGCCAGGGATTACCAC |
| U6 | FP-CGCTTCGGCAGCACATATACTAA RP-TATGGAACGCTTCACGAATTTGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, R.; Kanthaje, S.; Taneja, S.; Dhiman, R.K.; Chakraborti, A. miR-23b-3p Modulating Cytoprotective Autophagy and Glutamine Addiction in Sorafenib Resistant HepG2, a Hepatocellular Carcinoma Cell Line. Genes 2022, 13, 1375. https://doi.org/10.3390/genes13081375
Kaur R, Kanthaje S, Taneja S, Dhiman RK, Chakraborti A. miR-23b-3p Modulating Cytoprotective Autophagy and Glutamine Addiction in Sorafenib Resistant HepG2, a Hepatocellular Carcinoma Cell Line. Genes. 2022; 13(8):1375. https://doi.org/10.3390/genes13081375
Chicago/Turabian StyleKaur, Ramanpreet, Shruthi Kanthaje, Sunil Taneja, Radha K. Dhiman, and Anuradha Chakraborti. 2022. "miR-23b-3p Modulating Cytoprotective Autophagy and Glutamine Addiction in Sorafenib Resistant HepG2, a Hepatocellular Carcinoma Cell Line" Genes 13, no. 8: 1375. https://doi.org/10.3390/genes13081375