
Phylotranscriptomic and Evolutionary Analyses of the Green Algal Order Chaetophorales (Chlorophyceae, Chlorophyta)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture Conditions
2.2. Library Preparation and Sequencing
2.3. Quality Control, De Novo Assembly, and Sequence Annotation
2.4. Orthologous Group Identification and Phylotranscriptomic Analysis
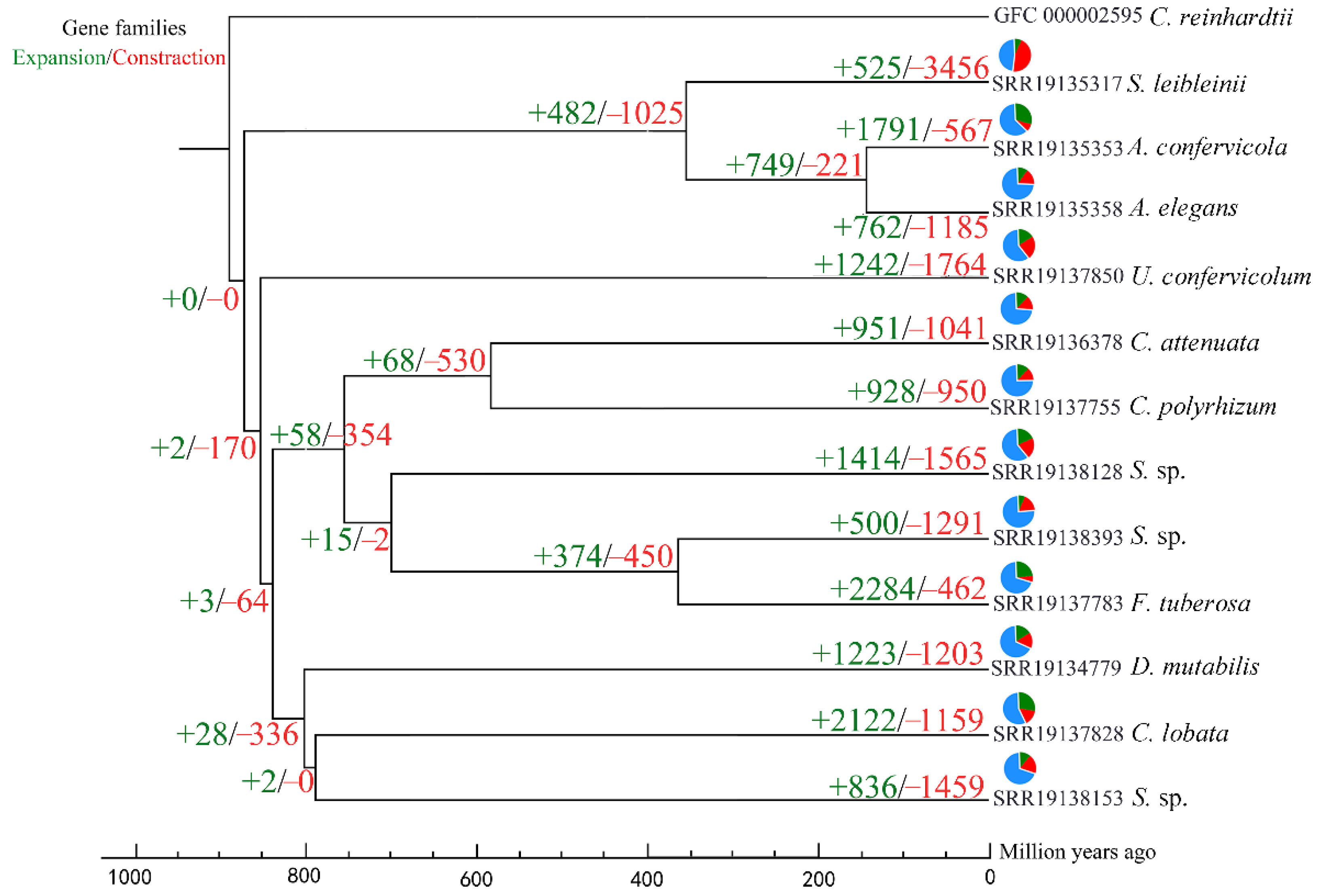
2.5. Gene Family Expansion and Contraction Analysis
2.6. Divergence Time Estimation
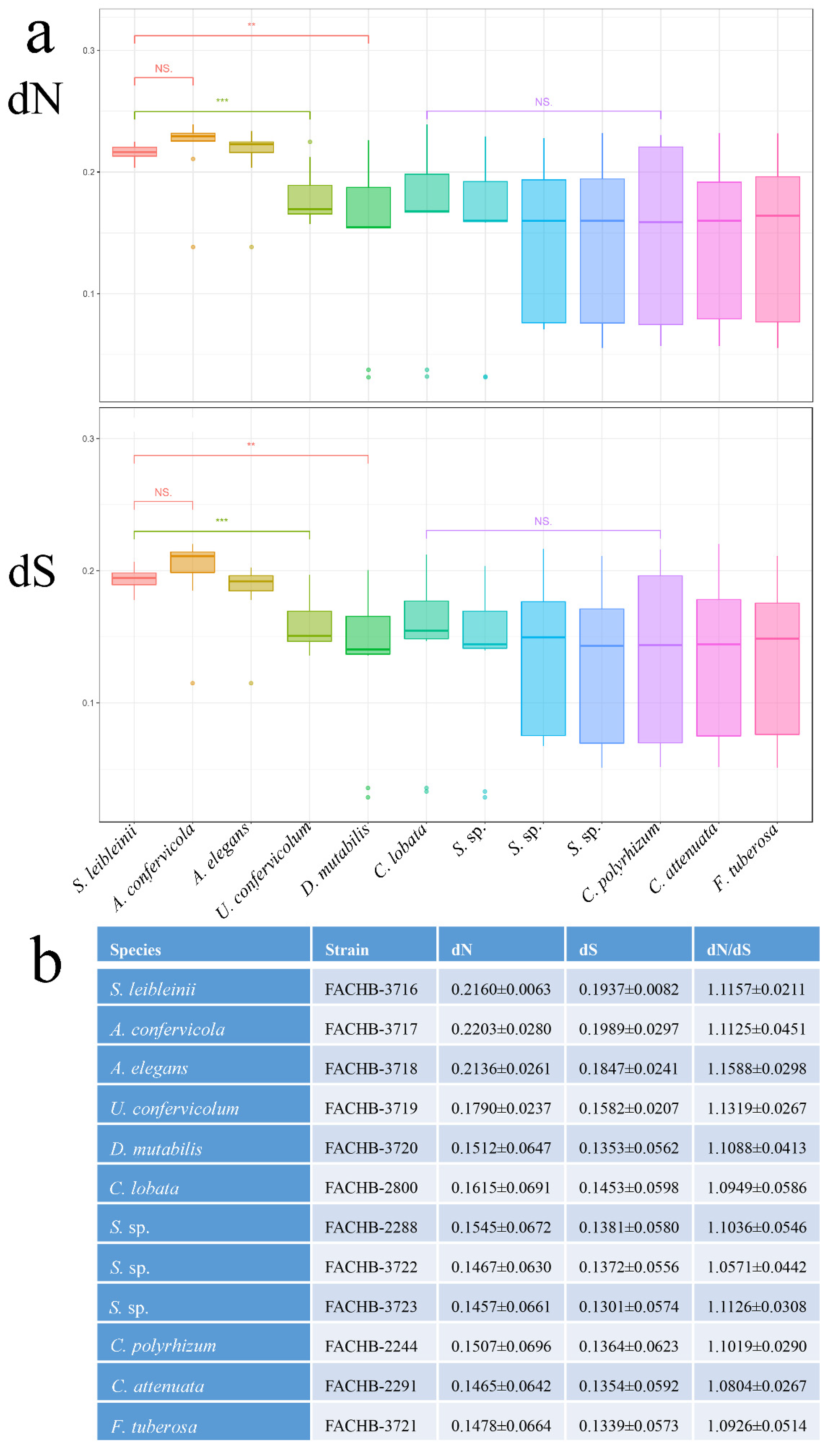
2.7. Substitution Rate Estimation
3. Results
3.1. De Novo Transcriptome Assembly and Orthology Detection
3.2. Phylotranscriptomic Analysis
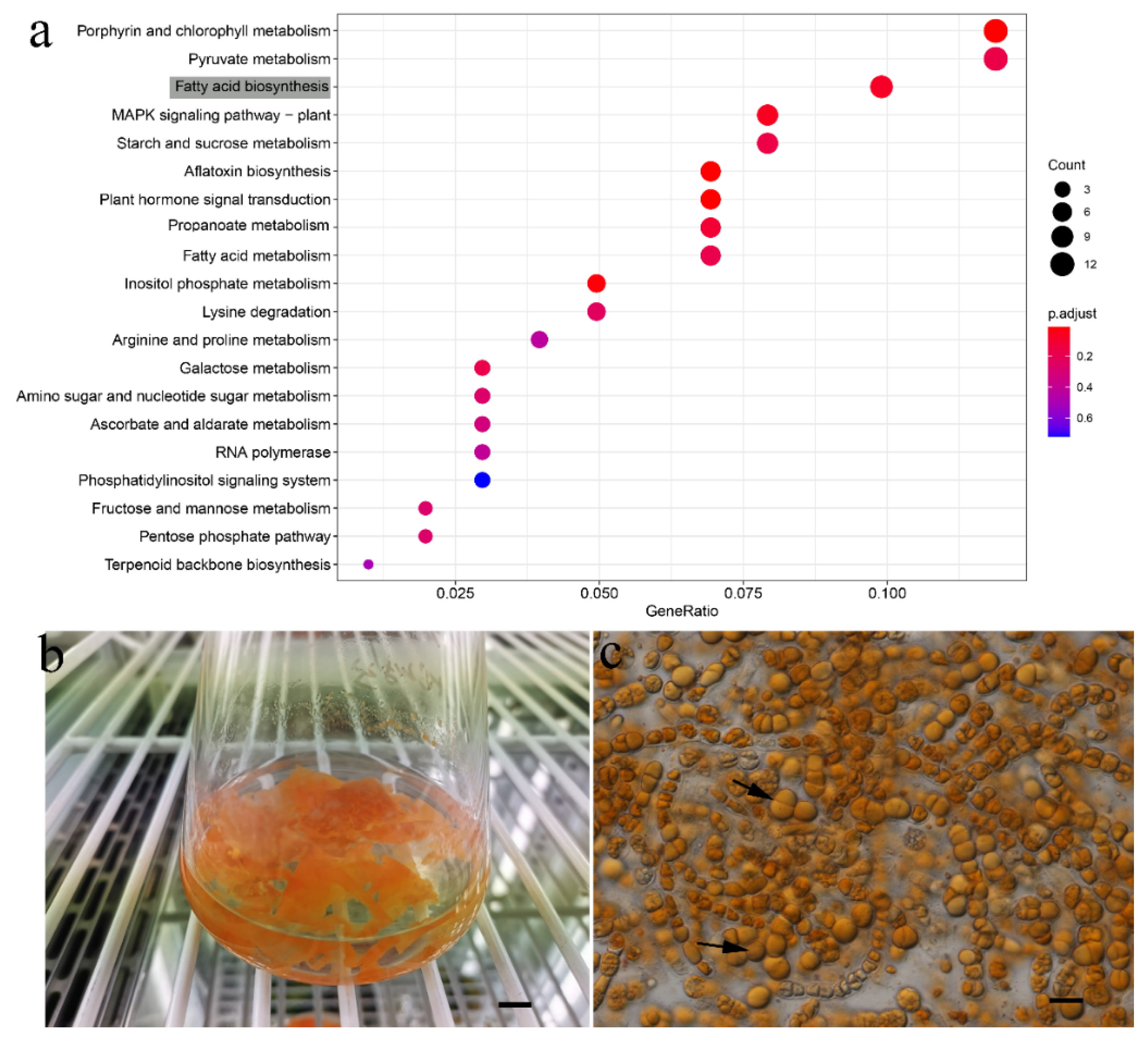
3.3. Gene Family Expansion and Contraction Analysis
3.4. Divergence Time Estimation
3.5. Substitution Rate Estimation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harding, J.P.C.; Whitton, B.A. Resistance to zinc of Stigeoclonium tenue in the field and the laboratory. Br. Phycol. J. 1976, 11, 417–426. [Google Scholar] [CrossRef]
- Özer, A.; Özer, D.; Ekiz, H.I. Application of Freundlich and Langmuir models to multistage purification process to remove heavy metal ions by using Schizomeris leibleinii. Process Biochem. 1999, 34, 919–927. [Google Scholar] [CrossRef]
- Pawlik-Skowrońska, B. Phytochelatin production in freshwater algae Stigeoclonium in response to heavy metals contained in mining water; effects of some environmental factors. Aquat. Toxicol. 2001, 52, 241–249. [Google Scholar] [CrossRef]
- Liu, J.Z.; Vyverman, W. Differences in nutrient uptake capacity of the benthic filamentous algae Cladophora sp., Klebsormidium sp. and Pseudanabaena sp. under varying N/P conditions. Bioresour. Technol. 2015, 179, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Danneels, B.; Vanormelingen, P.; Vyvermanb, W. Nutrient removal from horticultural waste water benthic filamentous algae Klebsormidium sp. Stigeoclonium spp. and their communities: From laboratory flask to outdoor Algal Turf Scrubber (ATS). Water Res. 2016, 92, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Vanormelingen, P.; Vyverman, W. Fatty acid profiles of four filamentous green algae under varying culture conditions. Bioresour. Technol. 2016, 200, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Caisová, L.; Marin, B.; Sausen, N.; Pröschold, T.; Melkonian, M. Polyphyly of Chaetophora and Stigeoclonium within the Chaetophorales (Chlorophyceae), revealed by sequence comparisons of nuclear-encoded SSU rRNA genes. J. Phycol. 2011, 47, 164–177. [Google Scholar] [CrossRef]
- Caisová, L.; Pérez Reyes, C.; Cruz Álamo, V.; Martel Quintana, A.; Surek, B.; Melkonian, M. Barrancaceae: A new green algal lineage with structural and behavioral adaptations to a fluctuating environment. Am. J. Bot. 2015, 102, 1482–1492. [Google Scholar] [CrossRef]
- Booton, G.C.; Floyd, G.L.; Fuerst, P.A. Origins and affinities of the filamentous green algal orders Chaetophorales and Oedogoniales based on 18SrRNA gene sequences. J. Phycol. 1998, 34, 312–318. [Google Scholar] [CrossRef]
- Buchheim, M.A.; Michalopulos, E.A.; Buchheim, J.A. Phylogeny of the Chlorophyceae with special reference to the Sphaeropleales: A study of 18S and 26S rRNA data. J. Phycol. 2001, 37, 819–835. [Google Scholar] [CrossRef]
- Turmel, M.; Brouard, J.S.; Gagnon, C.; Otis, C.; Lemieux, C. Deep division in the Chlorophyceae (Chlorophyta) revealed by chloroplast phylogenomic analyses. J. Phycol. 2008, 44, 739–750. [Google Scholar] [CrossRef]
- Caisová, L.; Melkonian, M. The Chaetophorales (Chlorophyceae)—A taxonomic revision at family level. Eur. J. Phycol. 2018, 53, 381–392. [Google Scholar] [CrossRef]
- Liu, B.W.; Hu, Y.X.; Hu, Z.Y.; Liu, G.X.; Zhu, H. Taxonomic scheme of the order Chaetophorales (Chlorophyceae, Chlorophyta) based on chloroplast genomes. BMC Genom. 2020, 21, 442. [Google Scholar] [CrossRef]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, S.; Barker, M.S.; Burleigh, J.G.; Gitzendanner, M.A.; et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 2014, 111, E4859–E4868. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.; Ma, X.; Shi, X.; Li, X.; Yang, L.; Xiao, S.; De Clerck, O.; Leliaert, F.; Zhong, B. Phylotranscriptomic insights into a Mesoproterozoic–Neoproterozoic origin and early radiation of green seaweeds (Ulvophyceae). Nat. Commun. 2022, 13, 1610. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.; Salomaki, E.D.; Lane, C.E.; Saunders, G.W. Kelp transcriptomes provide robust support for interfamilial relationships and revision of the little known Arthrothamnaceae (Laminariales). J. Phycol. 2017, 53, 1–6. [Google Scholar] [CrossRef]
- Zhang, Z.; Qu, C.; Yao, R.; Nie, Y.; Xu, C.; Miao, J.; Zhong, B. The parallel molecular adaptations to the Antarctic cold environment in two psychrophilic green algae. Genome. Biol. Evol. 2019, 11, 1897–1908. [Google Scholar] [CrossRef]
- Hu, Y.; Xing, W.; Song, H.; Hu, Z.; Liu, G. Comparison of colonial volvocine algae based on phylotranscriptomic analysis of gene family evolution and natural selection. Eur. J. Phycol. 2020, 55, 100–112. [Google Scholar] [CrossRef]
- Cheon, S.; Lee, S.; Hong, H.; Lee, H.; Kim, K.; Park, C. A guide to phylotranscriptomic analysis for phycologists. Algae 2021, 36, 333–340. [Google Scholar] [CrossRef]
- Xiong, Q.; Hu, Y.; Dong, X.; Chen, Y.; Liu, G.; Hu, Z. Phylotranscriptomic and Evolutionary Analyses of Oedogoniales (Chlorophyceae, Chlorophyta). Diversity 2002, 14, 157. [Google Scholar] [CrossRef]
- Hahn, M.W.; Demuth, J.P.; Han, S.G. Accelerated rate of gene gain and loss in primates. Genetics 2007, 177, 1941–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochnik, S.E.; Umen, J.; Nedelcu, A.M.; Hallmann, A.; Miller, S.M.; Nishii, I.; Ferris, P.; Kuo, A.; Mitros, T.; Fritz-Laylin, L.K.; et al. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 2010, 329, 223–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanschen, E.R.; Marriage, T.N.; Ferris, P.J.; Hamaji, T.; Toyoda, A.; Fujiyama, A.; Neme, R.; Noguchi, H.; Minakuchi, Y.; Suzuki, M.; et al. The Gonium pectorale genome demonstrates co-option of cell cycle regulation during the evolution of multicellularity. Nat. Commun. 2016, 7, 11370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Cho, Y.S.; Kim, H.M.; Chung, O.; Kim, H.; Jho, S.; Seomum, H.; Kim, J.; Bang, W.Y.; Kim, C.; et al. Comparison of carnivore, omnivore, and herbivore mammalian genomes with a new leopard assembly. Genome Biol. 2016, 17, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Featherston, J.; Arakaki, Y.; Hanschen, E.R.; Ferris, P.J.; Michod, R.E.; Olson, B.J.; Nozaki, H.; Durand, P.M. The 4-celled Tetrabaena socialis nuclear genome reveals the essential components for genetic control of cell number at the origin of multicellularity in the volvocine lineage. Mol. Biol. Evol. 2017, 35, 855–870. [Google Scholar] [CrossRef]
- Lenski, R.E.; Ofria, C.; Pennock, R.T.; Adami, C. The evolutionary origin of complex features. Nature 2003, 423, 139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, H.W.; Bold, H.C. Phycological Studies IV. Some Soil Algae from Enchanted Rock and Related Algal Species; University of Texas Publication: Denton, TX, USA, 1963; pp. 1–95. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic. Acids. Res. 2013, 41, e121. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Mark, P.V.D.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [Green Version]
- Caisová, L.; Marin, B.; Melkonian, M. A consensus secondary structure of ITS2 in the Chlorophyta identified by phylogenetic reconstruction. Protist 2013, 164, 482–496. [Google Scholar] [CrossRef]
- Maddison, W.P. Gene trees in species trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Huynen, M.A.; Bork, P. Measuring genome evolution. Proc. Natl. Acad. Sci. USA 1998, 95, 5849–5856. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, W.F. Phylogenetic classification and the universal tree. Science 1999, 284, 2124–2128. [Google Scholar] [CrossRef]
- Degnan, J.H.; Rosenberg, N.A. Discordance of species trees with their most likely gene trees. PLoS Genet. 2006, 2, e68. [Google Scholar] [CrossRef] [Green Version]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef]
- Zou, J.; Yue, W.; Li, L.; Wang, X.; Lu, J.; Duan, B.; Liu, J. DNA barcoding of recently diversified tree species: A case study on spruces based on 20 DNA fragments from three different genomes. Trees 2016, 30, 959–969. [Google Scholar] [CrossRef]
- Gernandt, D.S.; Aguirre Dugua, X.; Vázquez-Lobo, A.; Willyard, A.; Moreno Letelier, A.; Pérez de la Rosa, J.A.; Pinero, D.; Liston, A. Multi-locus phylogenetics, lineage sorting, and reticulation in Pinus subsection Australes. Am. J. Bot. 2018, 105, 711–725. [Google Scholar] [CrossRef] [Green Version]
- Wolf, Y.I.; Rogozin, I.B.; Grishin, N.V.; Koonin, E.V. Genome trees and the tree of life. Trends Genet. 2002, 18, 472–479. [Google Scholar] [CrossRef]
- Mirarab, S. Species tree estimation using ASTRAL: Practical considerations. arXiv 2019. [Google Scholar] [CrossRef]
- Davidson, R.; Vachaspati, P.; Mirarab, S.; Warnow, T. Phylogenomic species tree estimation in the presence of incomplete lineage sorting and horizontal gene transfer. BMC Genom. 2015, 16, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swenson, M.S.; Warnow, T. ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 2014, 30, i541–i548. [Google Scholar] [CrossRef] [Green Version]
- Wortley, A.H.; Rudall, P.J.; Harris, D.J.; Scotland, R.W. How much data are needed to resolve a difficult phylogeny? Case study in Lamiales. Syst. Biol. 2005, 54, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Hillis, D.M. Approaches for assessing phylogenetic accuracy. Syst. Biol. 1995, 44, 3–16. [Google Scholar] [CrossRef]
- Ruvolo, M. Molecular phylogeny of the hominoids: Inferences from multiple independent DNA sequence data sets. Mol. Biol. Evol. 1997, 14, 248–265. [Google Scholar] [CrossRef] [Green Version]
- Sang, T. Utility of low-copy nuclear gene sequences in plant phylogenetics. Crit. Rev. Biochem. Mol. 2002, 37, 121–147. [Google Scholar] [CrossRef]
- Prachumwat, A.; Li, W.H. Gene number expansion and contraction in vertebrate genomes with respect to invertebrate genomes. Genome Res. 2008, 18, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.L. Gene family evolution in green plants with emphasis on the origination and evolution of Arabidopsis thaliana genes. Plant J. 2013, 73, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Albalat, R.; Cañestro, C. Evolution by gene loss. Nat. Rev. Genet. 2016, 17, 379. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, J.; Chi, Q.; Li, Y.; Wang, H.; Gong, Y.; Liu, G.; Hu, Z.; Han, D.; Hu, Q. Critical assessment of the filamentous green microalga Oedocladium carolinianum for astaxanthin and oil production. Algal Res. 2022, 61, 102599. [Google Scholar] [CrossRef]
- Qiu, H.; Price, D.C.; Yang, E.C.; Yoon, H.S.; Bhattacharya, D. Evidence of ancient genome reduction in red algae (Rhodophyta). J. Phycol. 2015, 51, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000; pp. 33–50. [Google Scholar]
- Teng, L.; Fan, X.; Xu, D.; Zhang, X.; Mock, T.; Ye, N. Identification of genes under positive selection reveals differences in evolutionary adaptation between brown-algal species. Front Plant Sci. 2017, 8, 1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Voucher | Culture | No. of Raw Reads | No. of Clean Reads | N50 Length | Single-Copy Orthologs | Complete BUSCOs (%) | Number of Coding Sequence |
|---|---|---|---|---|---|---|---|---|
| Schizomeris leibleinii | HB202102 | FACHB-3716 | 53,719,904 | 53,250,356 | 2530 | 1531 | 86.40 | 19298 |
| Aphanochaete confervicola | HB201725 | FACHB-3717 | 54,935,394 | 54,925,846 | 1461 | 1023 | 87.00 | 68361 |
| Aphanochaete elegans | HB201732 | FACHB-3718 | 54,067,336 | 54,055,980 | 1864 | 1360 | 84.10 | 42264 |
| Uronema confervicolum | LY201701 | FACHB-3719 | 58,939,480 | 58,861,738 | 1937 | 1495 | 85.40 | 41588 |
| Draparnaldia mulabilis | AES201713 | FACHB-3720 | 76,239,588 | 76,226,452 | 1614 | 1472 | 83.40 | 31263 |
| Chaetophora lobata | QH201901 | FACHB-2800 | 55,627,796 | 55,487,446 | 1544 | 1186 | 81.60 | 49304 |
| Stigeoclonium sp. | HB201635 | FACHB-2288 | 68,612,258 | 68,599,684 | 2055 | 1690 | 82.30 | 29126 |
| F. tuberosa | HB201823 | FACHB-3721 | 58,548,230 | 58,465,136 | 1657 | 1308 | 82.80 | 55524 |
| Chaetophoropsis polyrhizum | HB201646 | FACHB-2244 | 60,022,402 | 60,008,734 | 2090 | 1634 | 83.20 | 34764 |
| Stigeoclonium sp. | HB201648 | FACHB-3722 | 60,955,222 | 60,946,270 | 1946 | 1471 | 83.50 | 31683 |
| Stigeoclonium sp. | YN201601 | FACHB-3723 | 63,709,968 | 63,690,832 | 1990 | 1762 | 83.70 | 32184 |
| Chaetophoropsis attenuata | FHB201644 | FACHB-2291 | 87,268,606 | 87,168,808 | 1720 | 1621 | 81.80 | 34151 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Chen, Y.; Zhu, H.; Liu, G. Phylotranscriptomic and Evolutionary Analyses of the Green Algal Order Chaetophorales (Chlorophyceae, Chlorophyta). Genes 2022, 13, 1389. https://doi.org/10.3390/genes13081389
Liu B, Chen Y, Zhu H, Liu G. Phylotranscriptomic and Evolutionary Analyses of the Green Algal Order Chaetophorales (Chlorophyceae, Chlorophyta). Genes. 2022; 13(8):1389. https://doi.org/10.3390/genes13081389
Chicago/Turabian StyleLiu, Benwen, Yangliang Chen, Huan Zhu, and Guoxiang Liu. 2022. "Phylotranscriptomic and Evolutionary Analyses of the Green Algal Order Chaetophorales (Chlorophyceae, Chlorophyta)" Genes 13, no. 8: 1389. https://doi.org/10.3390/genes13081389