
Transcriptomic Immune Profiles Can Represent the Tumor Immune Microenvironment Related to the Tumor Budding Histology in Uterine Cervical Cancer

 , , , and
, , , and
Abstract
:1. Introduction
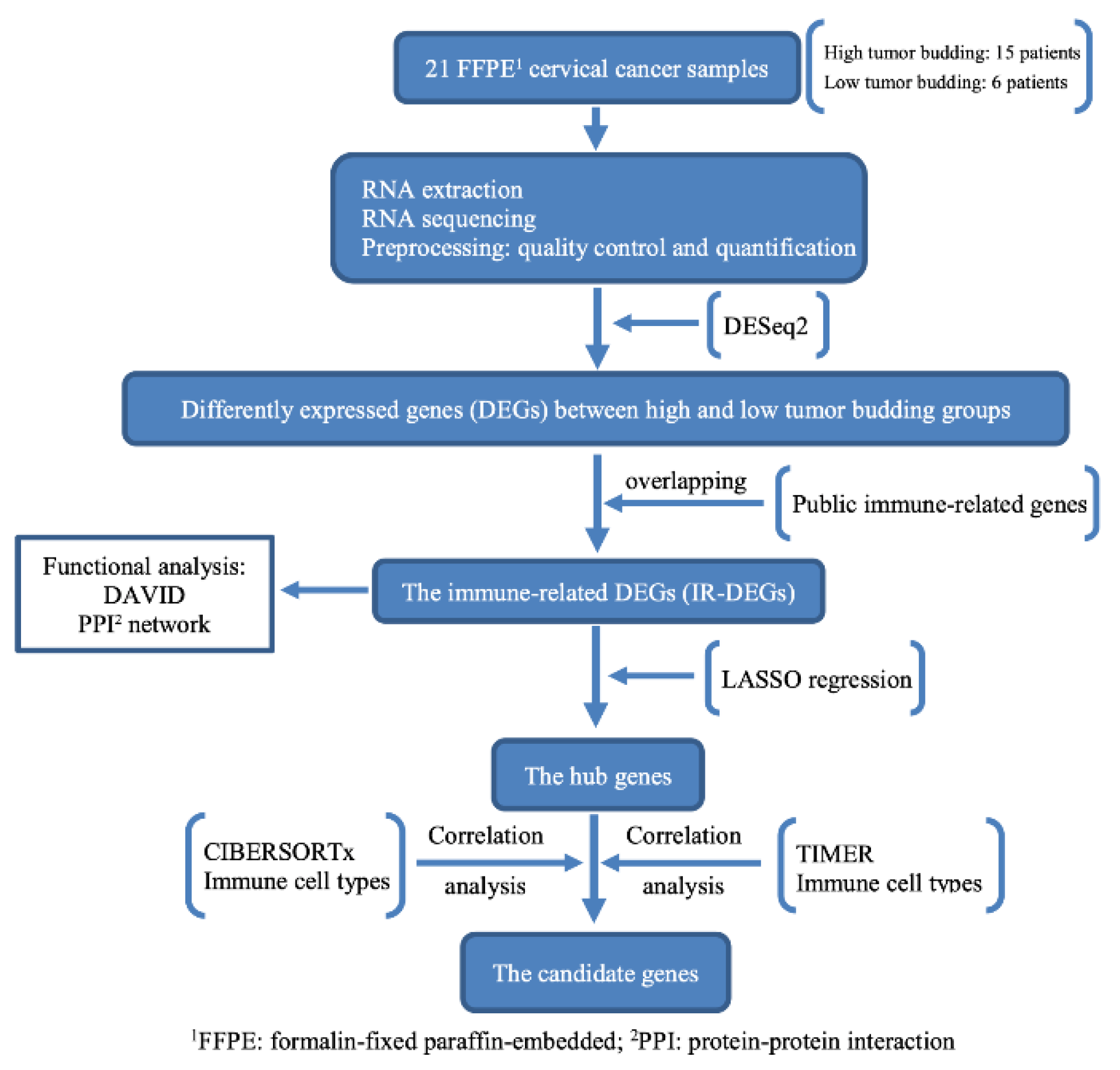
2. Materials and Methods
2.1. Sample Collection
2.2. Pathological Process
2.3. Clinical Parameters and Follow-Up
2.4. RNA Extraction and Sequencing
2.5. Bioinformatic Analysis
2.6. Immune-Related Gene Dataset
2.7. Functional Analysis
2.8. Immune-Cell-Type Analysis
2.9. Candidate Gene Analysis
2.10. Protein–Protein Interaction Network Analysis
2.11. Receiver Operating Characteristic (ROC) Curve and Survival Analysis
3. Results
3.1. Clinical Information
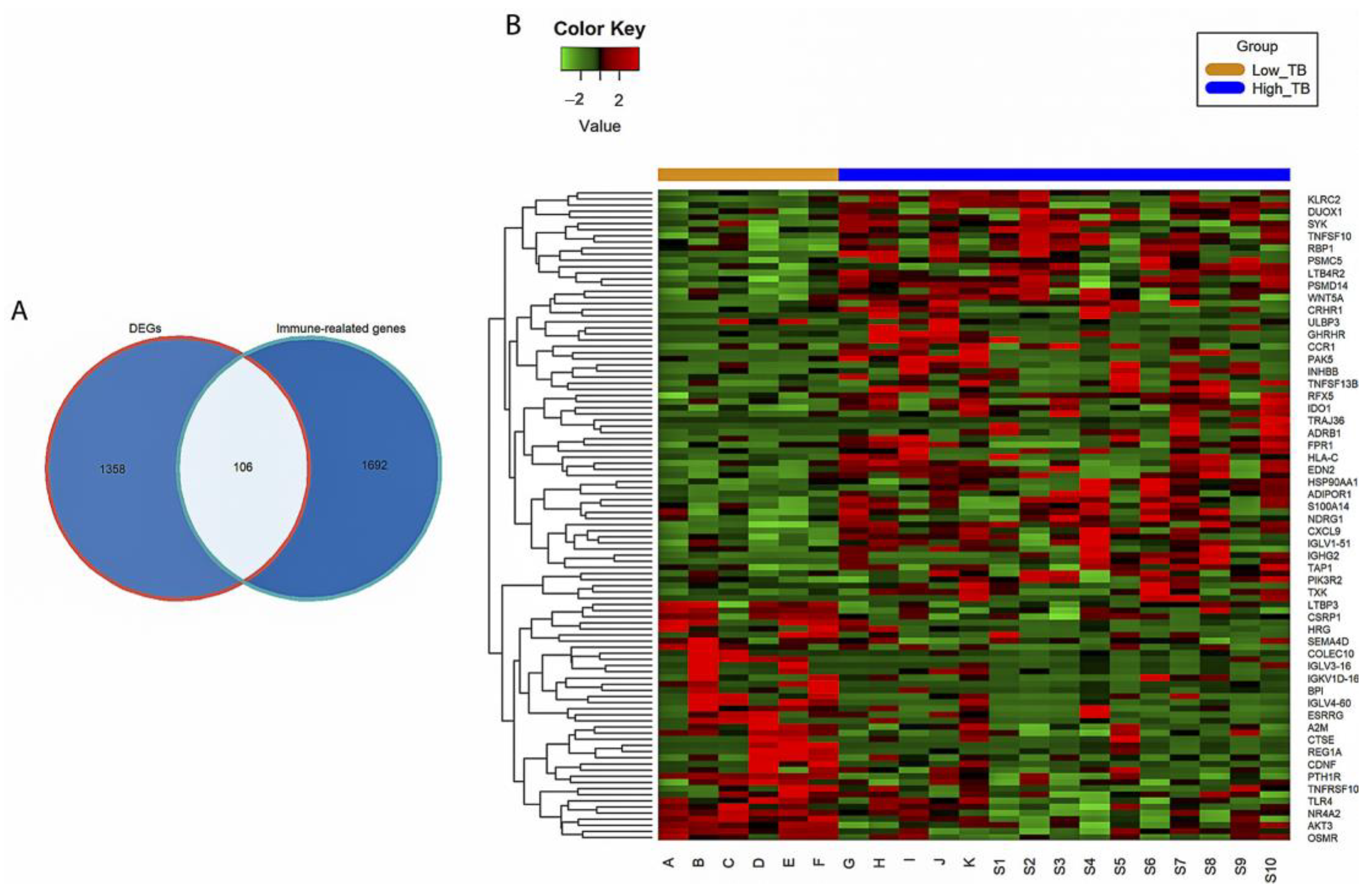
3.2. Identification of IR-DEGs
3.3. Functional Enrichment Analysis
3.4. Identification of Candidate Immune-Related Genes
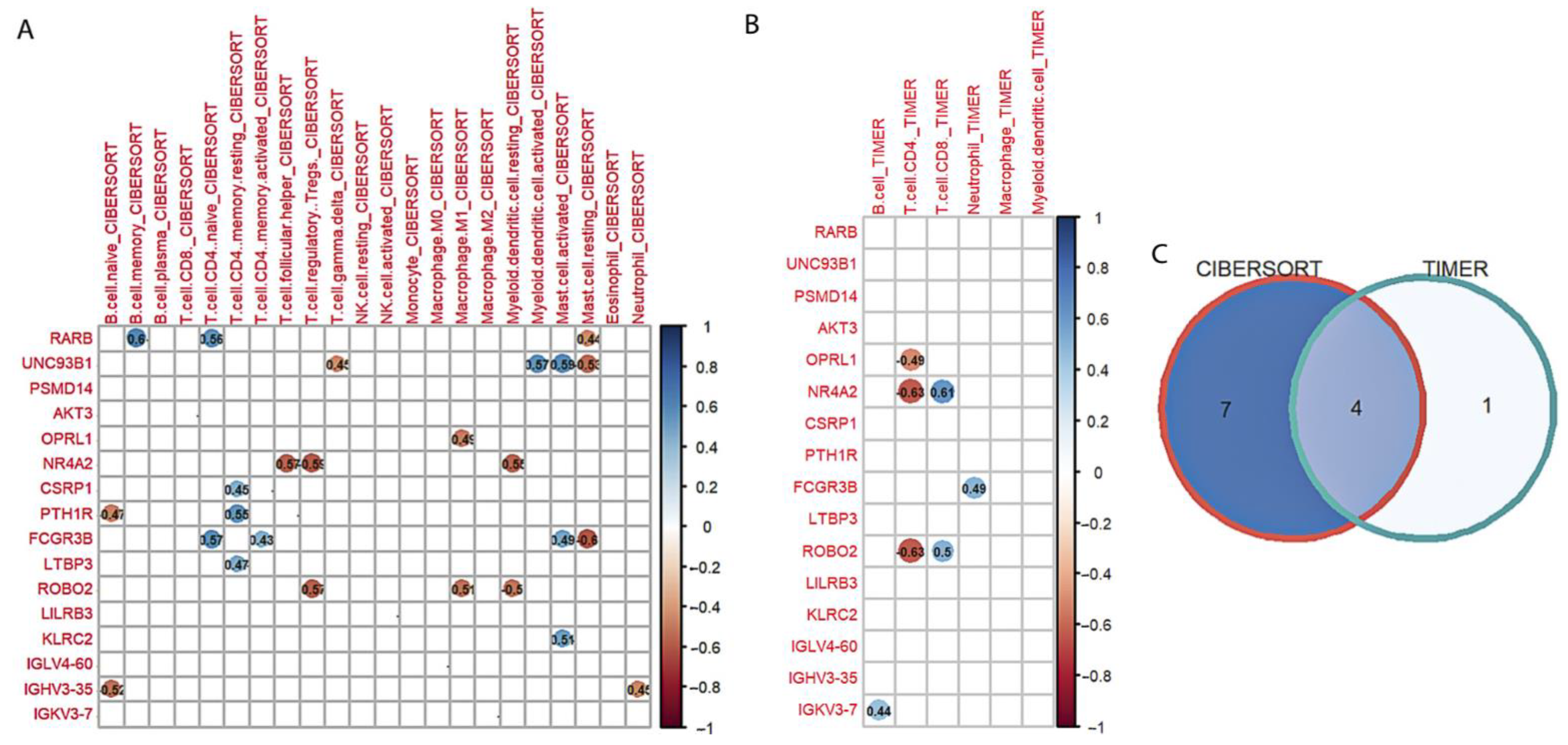
3.4.1. Selection of Hub Immune-Related Genes with LASSO Regression
3.4.2. Correlation Analysis
3.5. PPI Network Analysis
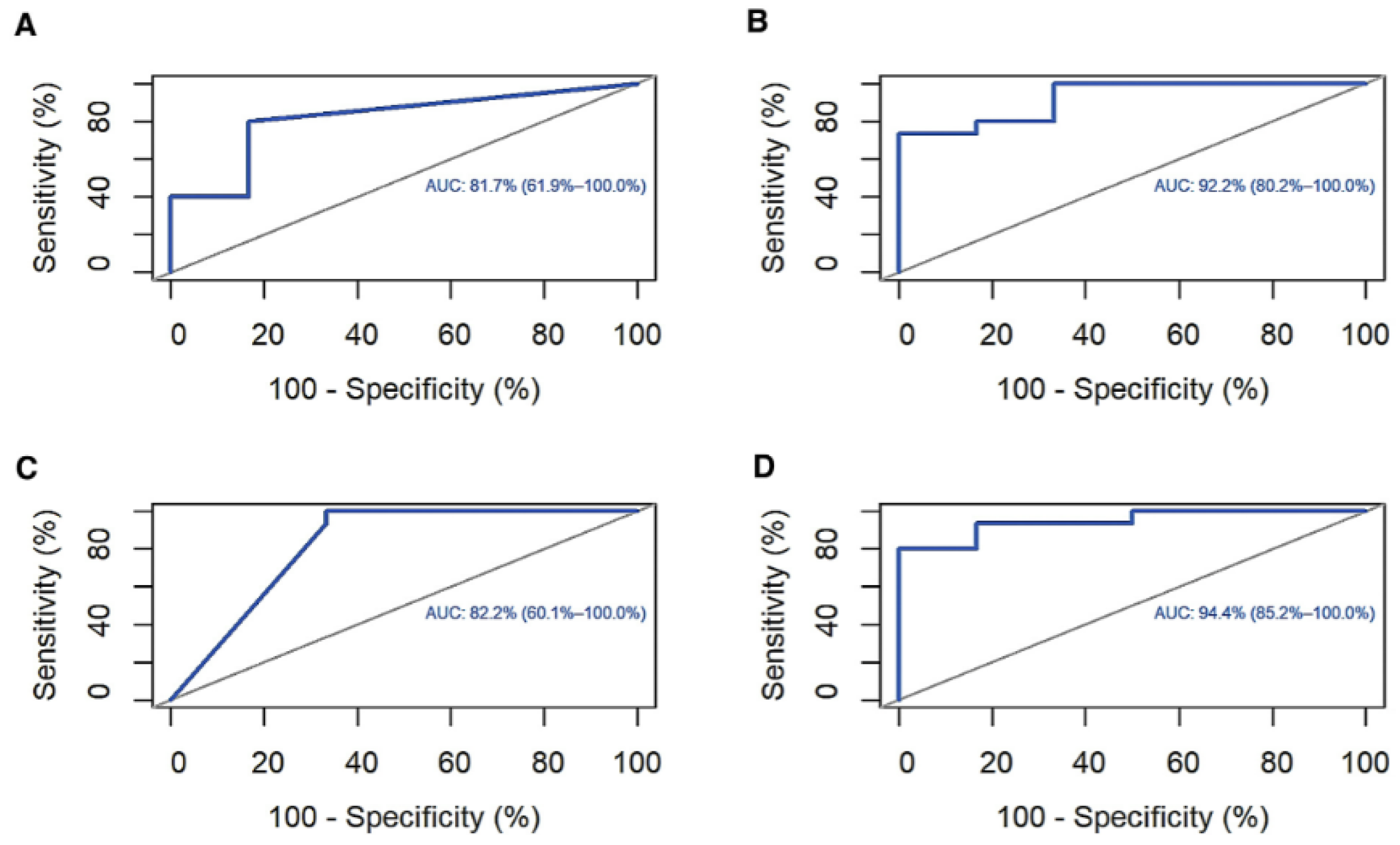
3.6. ROC Curve and Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020; Available online: https://gco.iarc.fr/today/about#references (accessed on 28 March 2022).
- Huang, B.; Cai, J.; Xu, X.; Guo, S.; Wang, Z. High-Grade Tumor Budding Stratifies Early-Stage Cervical Cancer with Recurrence Risk. PLoS ONE 2016, 11, e0166311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satabongkoch, N.; Khunamornpong, S.; Pongsuvareeyakul, T.; Settakorn, J.; Sukpan, K.; Soongkhaw, A.; Intaraphet, S.; Suprasert, P.; Siriaunkgul, S. Prognostic Value of Tumor Budding in Early-Stage Cervical Adenocarcinomas. Asian Pac. J. Cancer Prev. 2017, 18, 1717. [Google Scholar] [CrossRef] [PubMed]
- Chong, G.O.; Jee-Young Park, N.; Han, H.S.; Cho, J.; Kim, M.G.; Choi, Y.; Yeo, J.Y.; Lee, Y.H.; Hong, D.G.; Park, J.Y. Intratumoral Budding: A Novel Prognostic Biomarker for Tumor Recurrence and a Potential Predictor of Nodal Metastasis in Uterine Cervical Cancer. Eur. J. Surg. Oncol. 2021, 47, 3182–3187. [Google Scholar] [CrossRef]
- Lugli, A.; Zlobec, I.; Berger, M.D.; Kirsch, R.; Nagtegaal, I.D. Tumour Budding in Solid Cancers. Nat. Rev. Clin. Oncol. 2021, 18, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Ailia, M.J.; Thakur, N.; Chong, Y.; Yim, K. Tumor Budding in Gynecologic Cancer as a Marker for Poor Survival: A Systematic Review and Meta-Analysis of the Perspectives of Epithelial–Mesenchymal Transition. Cancers 2022, 14, 1431. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Chong, G.O.; Park, J.Y.; Chung, D.; Lee, Y.H.; Lee, H.J.; Hong, D.G.; Han, H.S.; Lee, Y.S. Tumor Budding in Cervical Cancer as a Prognostic Factor and Its Possible Role as an Additional Intermediate-Risk Factor. Gynecol. Oncol. 2020, 159, 157–163. [Google Scholar] [CrossRef]
- Jin, M.Z.; Jin, W.L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Schiavoni, G.; Gabriele, L.; Mattei, F. The Tumor Microenvironment: A Pitch for Multiple Players. Front. Oncol. 2013, 3, 90. [Google Scholar] [CrossRef] [Green Version]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Nearchou, I.P.; Lillard, K.; Gavriel, C.G.; Ueno, H.; Harrison, D.J.; Caie, P.D. Automated Analysis of Lymphocytic Infiltration, Tumor Budding, and Their Spatial Relationship Improves Prognostic Accuracy in Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, H.; Christe, L.; Eichmann, M.; Reinhard, S.; Zlobec, I.; Blank, A.; Lugli, A. Tumour Budding/T Cell Infiltrates in Colorectal Cancer: Proposal of a Novel Combined Score. Histopathology 2020, 76, 572–580. [Google Scholar] [CrossRef]
- Pecorelli, S.; Zigliani, L.; Odicino, F. Revised FIGO Staging for Carcinoma of the Cervix. Int. J. Gynecol. Obstet. 2009, 105, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Lugli, A.; Kirsch, R.; Ajioka, Y.; Bosman, F.; Cathomas, G.; Dawson, H.; El Zimaity, H.; Fléjou, J.F.; Hansen, T.P.; Hartmann, A.; et al. Recommendations for Reporting Tumor Budding in Colorectal Cancer Based on the International Tumor Budding Consensus Conference (ITBCC) 2016. Mod. Pathol. 2017, 30, 1299–1311. [Google Scholar] [CrossRef]
- Park, J.Y.; Hong, D.G.; Chong, G.O.; Park, J.Y. Tumor Budding Is a Valuable Diagnostic Parameter in Prediction of Disease Progression of Endometrial Endometrioid Carcinoma. Pathol. Oncol. Res. 2019, 25, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 September 2021).
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Bhattacharya, S.; Dunn, P.; Thomas, C.G.; Smith, B.; Schaefer, H.; Chen, J.; Hu, Z.; Zalocusky, K.A.; Shankar, R.D.; Shen-Orr, S.S.; et al. ImmPort, toward Repurposing of Open Access Immunological Assay Data for Translational and Clinical Research. Sci. Data 2018, 5, 180015. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive Analyses of Tumor Immunity: Implications for Cancer Immunotherapy. Genome Biol. 2016, 17, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, G.; Finotello, F.; Petitprez, F.; Zhang, J.D.; Baumbach, J.; Fridman, W.H.; List, M.; Aneichyk, T. Comprehensive Evaluation of Transcriptome-Based Cell-Type Quantification Methods for Immuno-Oncology. Bioinformatics 2019, 35, i436–i445. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots. R Package Version 0.4.0. 2020. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 10 July 2022).
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, T.; Simko, V. R Package “corrplot”: Visualization of a Correlation Matrix (Version 0.92). 2021. Available online: https://github.com/taiyun/corrplot (accessed on 10 July 2022).
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein–Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. PROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Kassambara, A.; Kosinski, M.; Biece, P. Survminer: Drawing Survival Curves Using “ggplot2”. R Package Version 0.4.9. 2021. Available online: https://CRAN.R-project.org/package=survminer (accessed on 10 July 2022).
- Therneau, T. A Package for Survival Analysis in R. R Package Version 3.3-1. 2022. Available online: https://CRAN.R-project.org/package=survival (accessed on 10 July 2022).
- Therneau, M.T.; Grambsch, M.P. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000; ISBN 978-1-4757-3294-8. [Google Scholar]
- Wartenberg, M.; Cibin, S.; Zlobec, I.; Vassella, E.; Eppenberger-Castori, S.; Terracciano, L.; Eichmann, M.D.; Worni, M.; Gloor, B.; Perren, A.; et al. Integrated Genomic and Immunophenotypic Classification of Pancreatic Cancer Reveals Three Distinct Subtypes with Prognostic/ Predictive Significance. Clin. Cancer Res. 2018, 24, 4444–4454. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Iglesias, T.; del Toro-Arreola, A.; Albarran-Somoza, B.; del Toro-Arreola, S.; Sanchez-Hernandez, P.E.; Ramirez-Dueñas, M.; Balderas-Peña, L.M.A.; Bravo-Cuellar, A.; Ortiz-Lazareno, P.C.; Daneri-Navarro, A. Low NKp30, NKp46 and NKG2D Expression and Reduced Cytotoxic Activity on NK Cells in Cervical Cancer and Precursor Lesions. BMC Cancer 2009, 9, 186. [Google Scholar] [CrossRef] [Green Version]
- Junker, F.; Gordon, J.; Qureshi, O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol. 2020, 11, 1393. [Google Scholar] [CrossRef]
- Patel, K.R.; Roberts, J.T.; Barb, A.W. Multiple Variables at the Leukocyte Cell Surface Impact Fc γ Receptor-Dependent Mechanisms. Front. Immunol. 2019, 10, 223. [Google Scholar] [CrossRef] [Green Version]
- Barb, A.W. Fc γ Receptor Compositional Heterogeneity: Considerations for Immunotherapy Development. J. Biol. Chem 2021, 296, 100057. [Google Scholar] [CrossRef]
- Treffers, L.W.; van Houdt, M.; Bruggeman, C.W.; Heineke, M.H.; Zhao, X.W.; van der Heijden, J.; Nagelkerke, S.Q.; Verkuijlen, P.J.J.H.; Geissler, J.; Lissenberg-Thunnissen, S.; et al. FcγRIIIb Restricts Antibody-Dependent Destruction of Cancer Cells by Human Neutrophils. Front. Immunol. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Palano, M.T.; Gallazzi, M.; Cucchiara, M.; de Lerma Barbaro, A.; Gallo, D.; Bassani, B.; Bruno, A.; Mortara, L. Neutrophil and Natural Killer Cell Interactions in Cancers: Dangerous Liaisons Instructing Immunosuppression and Angiogenesis. Vaccines 2021, 9, 1488. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK Cells for Cancer Immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Bassani, B.; Baci, D.; Gallazzi, M.; Poggi, A.; Bruno, A.; Mortara, L. Natural Killer Cells as Key Players of Tumor Progression and Angiogenesis: Old and Novel Tools to Divert Their Pro-Tumor Activities into Potent Anti-Tumor Effects. Cancers 2019, 11, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehlen, P.; Delloye-Bourgeois, C.; Chédotal, A. Novel Roles for Slits and Netrins: Axon Guidance Cues as Anticancer Targets? Nat. Rev. Cancer 2011, 11, 188–197. [Google Scholar] [CrossRef]
- Tessier-Lavigne, M.; Goodman, C.S. The Molecular Biology of Axon Guidance. J. S. Nye R. Kopan Curro Bioi 1995, 210, 27. [Google Scholar] [CrossRef] [Green Version]
- Kidd, T.; Bland, K.S.; Goodman, C.S. Slit Is the Midline Repellent for the Robo Receptor in Drosophila. Cell 1999, 96, 785–794. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; Mazumder-Indra, D.; Mondal, R.K.; Basu, P.S.; Roy, A.; Roychoudhury, S.; Panda, C.K. Inactivation of SLIT2-ROBO1/2 Pathway in Premalignant Lesions of Uterine Cervix: Clinical and Prognostic Significances. PLoS ONE 2012, 7, e38342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, M.S.; Hinck, L. A Roundabout Way to Cancer. Adv. Cancer Res. 2012, 114, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhosle, V.K.; Mukherjee, T.; Huang, Y.W.; Patel, S.; Pang, B.W.F.; Liu, G.Y.; Glogauer, M.; Wu, J.Y.; Philpott, D.J.; Grinstein, S.; et al. SLIT2/ROBO1-Signaling Inhibits Macropinocytosis by Opposing Cortical Cytoskeletal Remodeling. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Inamoto, T.; Czerniak, B.A.; Dinney, C.P.; Kamat, A.M. Cytoplasmic Mislocalization of the Orphan Nuclear Receptor Nurr1 Is a Prognostic Factor in Bladder Cancer. Cancer 2010, 116, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.F.; Cao, G.W. Role of Nuclear Receptor NR4A2 in Gastrointestinal Inflammation and Cancers. World J. Gastroenterol. 2012, 18, 6865. [Google Scholar] [CrossRef] [PubMed]
- Ke, N.; Claassen, G.; Yu, D.H.; Albers, A.; Fan, W.; Tan, P.; Grifman, M.; Hu, X.; DeFife, K.; Nguy, V.; et al. Nuclear Hormone Receptor NR4A2 Is Involved in Cell Transformation and Apoptosis. Cancer Res. 2004, 64, 8208–8212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, R.; Imai, A.; Mima, M.; Yamada, S.; Takeuchi, K.; Mochizuki, D.; Shinmura, D.; Kita, J.Y.; Nakagawa, T.; Kurokawa, T.; et al. Novel Prognostic Value and Potential Utility of Opioid Receptor Gene Methylation in Liquid Biopsy for Oral Cavity Cancer. Curr. Probl. Cancer 2022, 46, 100834. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. G Protein-Coupled Receptors: Novel Targets for Drug Discovery in Cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- Bedini, A.; Baiula, M.; Vincelli, G.; Formaggio, F.; Lombardi, S.; Caprini, M.; Spampinato, S. Nociceptin/Orphanin FQ Antagonizes Lipopolysaccharide-Stimulated Proliferation, Migration and Inflammatory Signaling in Human Glioblastoma U87 Cells. Biochem. Pharmacol. 2017, 140, 89–104. [Google Scholar] [CrossRef]
- Waits, P.S.; Purcell, W.M.; Fulford, A.J.; McLeod, J.D. Nociceptin/Orphanin FQ Modulates Human T Cell Function in Vitro. J. Neuroimmunol. 2004, 149, 110–120. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (ALL) | N | |
|---|---|---|
| N = 21 | ||
| Age (range) | 48 (32–71) | 21 |
| Clinical Stage: | 21 | |
| Early stage | 17 (81.0%) | |
| Late stage | 4 (19.0%) | |
| Histology: | 21 | |
| Squamous cell carcinoma | 15 (71.4%) | |
| Adenocarcinoma | 5 (23.8%) | |
| Adenosquamous cell carcinoma | 1 (4.8%) | |
| Tumor budding: | 21 | |
| Low (<5 TBs 1) | 6 (28.6%) | |
| High (≥5 TBs) | 15 (71.4%) |
| Term Name | Description | Genes | FDR |
|---|---|---|---|
| hsa04650 | Natural killer cell mediated cytotoxicity | 11 | 9.34 × 10−9 |
| hsa04080 | Neuroactive ligand-receptor interaction | 12 | 4.44 × 10−6 |
| hsa05152 | Tuberculosis | 8 | 5.28 × 10−5 |
| hsa04380 | Osteoclast differentiation | 7 | 6.87 × 10−5 |
| hsa04360 | Axon guidance | 7 | 4.20 × 10−4 |
| hsa04145 | Phagosome | 6 | 9.10 × 10−4 |
| hsa05150 | Staphylococcus aureus infection | 5 | 9.10 × 10−4 |
| Category | Term Name | Description | Count | FDR |
|---|---|---|---|---|
| GO–BP 1 | GO:0007165 | Signal transduction | 73 | 1.60 × 10−23 |
| GO–BP | GO:0006955 | Immune response | 42 | 2.38 × 10−18 |
| GO–MF 2 | GO:0005102 | Signaling receptor binding | 41 | 2.81 × 10−17 |
| GO–MF | GO:0038023 | Signaling receptor activity | 36 | 3.54 × 10−14 |
| GO–BP | GO:0050776 | Regulation of immune response | 27 | 4.30 × 10−12 |
| GO–BP | GO:0048584 | Positive regulation of response to stimulus | 40 | 8.52 × 10−12 |
| GO–CC 3 | GO:0009986 | Cell surface | 26 | 1.49 × 10−11 |
| GO–BP | GO:0006935 | Chemotaxis | 20 | 5.35 × 10−10 |
| GO–MF | GO:0008528 | G protein-coupled peptide receptor activity | 12 | 6.22 × 10−09 |
| GO–BP | GO:0051239 | Regulation of multicellular organismal process | 42 | 2.67 × 10−08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, T.M.; Nguyen, H.D.T.; Lee, E.; Lee, D.; Choi, Y.S.; Cho, J.; Park, N.J.-Y.; Han, H.S.; Chong, G.O. Transcriptomic Immune Profiles Can Represent the Tumor Immune Microenvironment Related to the Tumor Budding Histology in Uterine Cervical Cancer. Genes 2022, 13, 1405. https://doi.org/10.3390/genes13081405
Le TM, Nguyen HDT, Lee E, Lee D, Choi YS, Cho J, Park NJ-Y, Han HS, Chong GO. Transcriptomic Immune Profiles Can Represent the Tumor Immune Microenvironment Related to the Tumor Budding Histology in Uterine Cervical Cancer. Genes. 2022; 13(8):1405. https://doi.org/10.3390/genes13081405
Chicago/Turabian StyleLe, Tan Minh, Hong Duc Thi Nguyen, Eunmi Lee, Donghyeon Lee, Ye Seul Choi, Junghwan Cho, Nora Jee-Young Park, Hyung Soo Han, and Gun Oh Chong. 2022. "Transcriptomic Immune Profiles Can Represent the Tumor Immune Microenvironment Related to the Tumor Budding Histology in Uterine Cervical Cancer" Genes 13, no. 8: 1405. https://doi.org/10.3390/genes13081405