
A Novel Intragenic Duplication in the HDAC8 Gene Underlying a Case of Cornelia de Lange Syndrome

 , , , , ,
, , , , ,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Clinical Diagnosis
2.2. Isolation of DNA and RNA
2.3. Next-Generation Sequencing
2.4. Array Comparative Genomic Hybridization
2.5. cDNA Synthesis and Analysis
2.6. Real-Time Quantitative PCR (qPCR)
2.7. Structure Modeling of HDAC8 Variant and Molecular Dynamics Simulation
3. Results
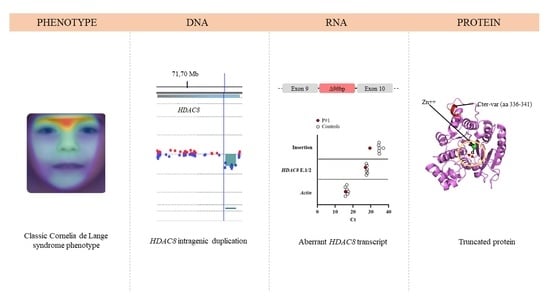
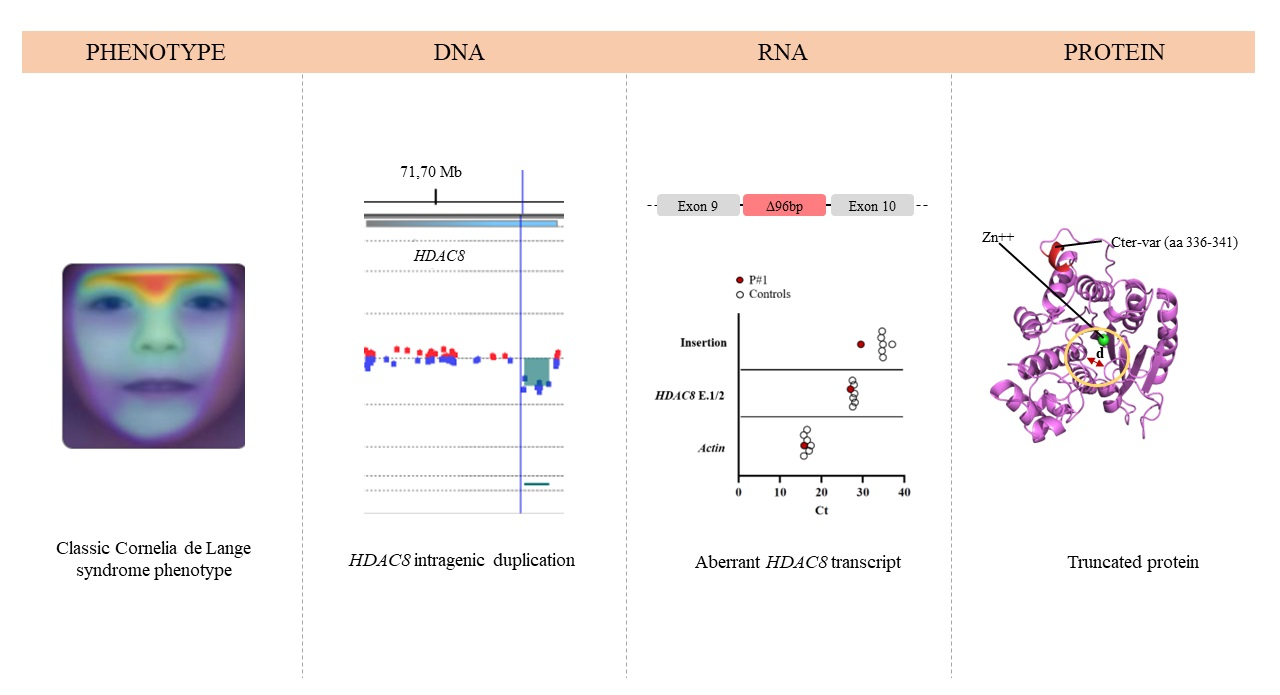
3.1. Clinical Report
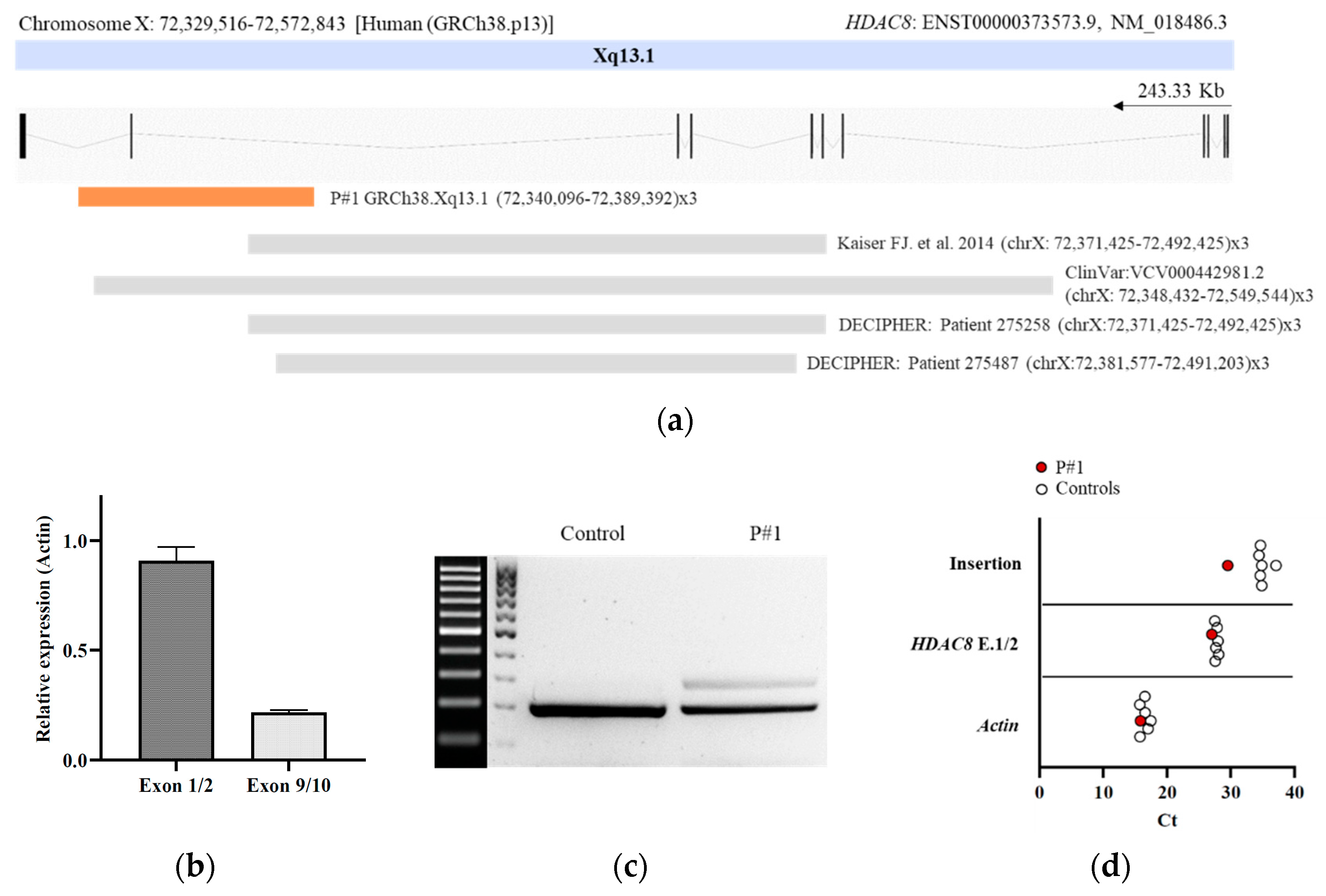
3.2. DNA Molecular Analyses
3.3. RNA Molecular Analyses
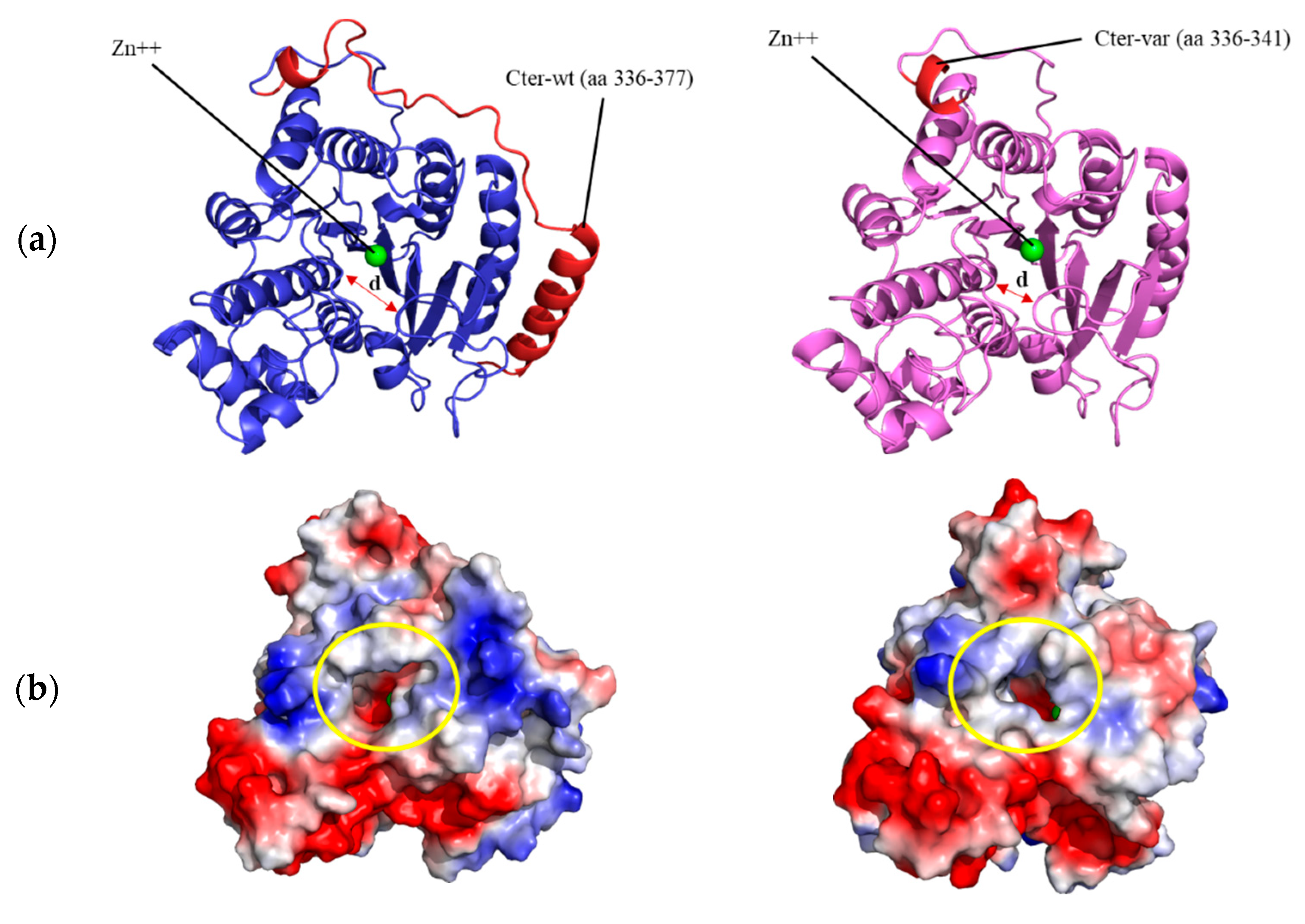
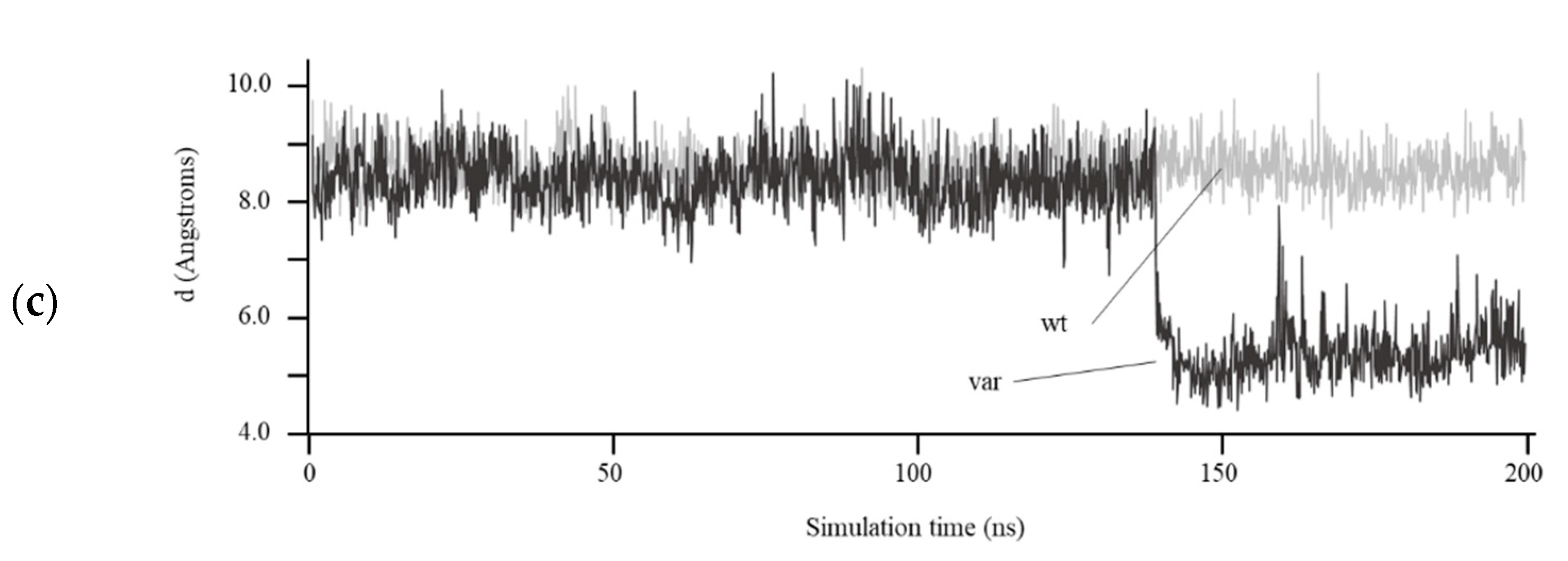
3.4. Structural Prediction of HDAC8 Variant
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Mannini, L.; Cucco, F.; Quarantotti, V.; Krantz, I.D.; Musio, A. Mutation Spectrum and Genotype-Phenotype Correlation in Cornelia de Lange Syndrome. Hum. Mutat. 2013, 34, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Krab, L.C.; Marcos-Alcalde, I.; Assaf, M.; Balasubramanian, M.; Andersen, J.B.; Bisgaard, A.-M.; Fitzpatrick, D.R.; Gudmundsson, S.; Huisman, S.A.; Kalayci, T.; et al. Delineation of phenotypes and genotypes related to cohesin structural protein RAD21. Hum. Genet. 2020, 139, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes and genotypes in individuals with SMC1A variants. Am. J. Med. Genet. A 2017, 173, 2108–2125. [Google Scholar] [CrossRef] [PubMed]
- Gil-Rodríguez, M.C.; Deardorff, M.A.; Ansari, M.; Tan, C.A.; Parenti, I.; Baquero-Montoya, C.; Ousager, L.B.; Puisac, B.; Hernández-Marcos, M.; Teresa-Rodrigo, M.E.; et al. De Novo Heterozygous Mutations in SMC3 Cause a Range of Cornelia de Lange Syndrome-Overlapping Phenotypes. Hum. Mutat. 2015, 36, 454–462. [Google Scholar] [CrossRef]
- Kaiser, F.J.; Ansari, M.; Braunholz, D.; Gil-Rodríguez, M.C.; Decroos, C.; Wilde, J.J.; Fincher, C.T.; Kaur, M.; Bando, M.; Amor, D.J.; et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum. Mol. Genet. 2014, 23, 2888–2900. [Google Scholar] [CrossRef]
- Parenti, I.; Gervasini, C.; Pozojevic, J.; Wendt, K.S.; Watrin, E.; Azzollini, J.; Braunholz, D.; Buiting, K.; Cereda, A.; Engels, H.; et al. Expanding the clinical spectrum of the ‘HDAC8 -phenotype’—Implications for molecular diagnostics, counseling and risk prediction: Expanding the clinical spectrum of the ‘HDAC8 -phenotype’. Clin. Genet. 2016, 89, 564–573. [Google Scholar] [CrossRef]
- Latorre-Pellicer, A.; Ascaso, Á.; Lucia-Campos, C.; Gil-Salvador, M.; Arnedo, M.; Antoñanzas, R.; Ayerza-Casas, A.; Marcos-Alcalde, I.; Gómez-Puertas, P.; Ramos, F.J.; et al. Things are not always what they seem: From Cornelia de Lange to KBG phenotype in a girl with genetic variants in NIPBL and ANKRD11. Mol. Genet. Genom. Med. 2021, 9, e1826. [Google Scholar] [CrossRef]
- Latorre-Pellicer, A.; Gil-Salvador, M.; Parenti, I.; Lucia-Campos, C.; Trujillano, L.; Marcos-Alcalde, I.; Arnedo, M.; Ascaso, Á.; Ayerza-Casas, A.; Antoñanzas-Pérez, R.; et al. Clinical relevance of postzygotic mosaicism in Cornelia de Lange syndrome and purifying selection of NIPBL variants in blood. Sci. Rep. 2021, 11, 15459. [Google Scholar] [CrossRef]
- Russo, S.; Masciadri, M.; Gervasini, C.; Azzollini, J.; Cereda, A.; Zampino, G.; Haas, O.; Scarano, G.; Di Rocco, M.; Finelli, P.; et al. Intragenic and large NIPBL rearrangements revealed by MLPA in Cornelia de Lange patients. Eur. J. Hum. Genet. 2012, 20, 734–741. [Google Scholar] [CrossRef][Green Version]
- Baquero-Montoya, C.; Gil-Rodríguez, M.C.; Teresa-Rodrigo, M.E.; Hernández-Marcos, M.; Bueno-Lozano, G.; Bueno-Martínez, I.; Remeseiro, S.; Fernández-Hernández, R.; Bassecourt-Serra, M.; de Alba, M.R.; et al. Could a patient with SMC1A duplication be classified as a human cohesinopathy?: Could a patient with SMC1A duplication be classified as a human cohesinopathy? Clin. Genet. 2014, 85, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Helgeson, M.; Keller-Ramey, J.; Knight Johnson, A.; Lee, J.A.; Magner, D.B.; Deml, B.; Deml, J.; Hu, Y.-Y.; Li, Z.; Donato, K.; et al. Molecular characterization of HDAC8 deletions in individuals with atypical Cornelia de Lange syndrome. J. Hum. Genet. 2018, 63, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zhang, N.; Pati, D. Cohesin subunit RAD21: From biology to disease. Gene 2020, 758, 144966. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pellicer, A.; Ascaso, Á.; Trujillano, L.; Gil-Salvador, M.; Arnedo, M.; Lucia-Campos, C.; Antoñanzas-Pérez, R.; Marcos-Alcalde, I.; Parenti, I.; Bueno-Lozano, G.; et al. Evaluating Face2Gene as a Tool to Identify Cornelia de Lange Syndrome by Facial Phenotypes. Int. J. Mol. Sci. 2020, 21, 1042. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural Snapshots of Human HDAC8 Provide Insights into the Class I Histone Deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef]
- Marcos-Alcalde, Í.; Mendieta-Moreno, J.I.; Puisac, B.; Gil-Rodríguez, M.C.; Hernández-Marcos, M.; Soler-Polo, D.; Ramos, F.J.; Ortega, J.; Pie, J.; Mendieta, J.; et al. Two-step ATP-driven opening of cohesin head. Sci. Rep. 2017, 7, 3266. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef]
- Sun, L. A commentary on exome sequencing identifies a de novo mutation in HDAC8 associated with Cornelia de Lange syndrome. J. Hum. Genet. 2014, 59, 479. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhou, D.; Zhang, Z.; Liu, Y.; Yang, Y. Exome sequencing identifies a de novo mutation in HDAC8 associated with Cornelia de Lange syndrome. J. Hum. Genet. 2015, 60, 165. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Huang, Z.; Fan, Y.; Sun, Y.; Liu, H.; Wang, L.; Gu, X.-F.; Yu, Y. A Functional Mutation in HDAC8 Gene as Novel Diagnostic Marker for Cornelia De Lange Syndrome. Cell. Physiol. Biochem. 2018, 47, 2388–2395. [Google Scholar] [CrossRef] [PubMed]
- Jezela-Stanek, A.; Murcia, P.V.; Jurkiewicz, D.; Iwanicka-Pronicka, K.; Jędrzejowska, M.; Krajewska-Walasek, M.; Płoski, R. Novel variant in HDAC8 gene resulting in the severe Cornelia de Lange phenotype. Clin. Dysmorphol. 2019, 28, 124–128. [Google Scholar] [CrossRef]
- Mio, C.; Passon, N.; Fogolari, F.; Cesario, C.; Novelli, A.; Pittini, C.; Damante, G. A novel de novo HDAC8 missense mutation causing Cornelia de Lange syndrome. Mol. Genet. Genom. Med. 2021, 9, e1612. [Google Scholar] [CrossRef]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef]
- Decroos, C.; Bowman, C.M.; Moser, J.A.S.; Christianson, K.E.; Deardorff, M.A.; Christianson, D.W. Compromised Structure and Function of HDAC8 Mutants Identified in Cornelia de Lange Syndrome Spectrum Disorders. ACS Chem. Biol. 2014, 9, 2157–2164. [Google Scholar] [CrossRef]
- Decroos, C.; Christianson, N.H.; Gullett, L.E.; Bowman, C.M.; Christianson, K.E.; Deardorff, M.A.; Christianson, D.W. Biochemical and Structural Characterization of HDAC8 Mutants Associated with Cornelia de Lange Syndrome Spectrum Disorders. Biochemistry 2015, 54, 6501–6513. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Porter, N.J.; Christianson, D.W. Structural aspects of HDAC8 mechanism and dysfunction in Cornelia de Lange syndrome spectrum disorders: Structural Aspects of HDAC8 Mechanism. Protein Sci. 2016, 25, 1965–1976. [Google Scholar] [CrossRef]
- Osko, J.D.; Porter, N.J.; Decroos, C.; Lee, M.S.; Watson, P.R.; Raible, S.E.; Krantz, I.D.; Deardorff, M.A.; Christianson, D.W. Structural analysis of histone deacetylase 8 mutants associated with Cornelia de Lange Syndrome spectrum disorders. J. Struct. Biol. 2021, 213, 107681. [Google Scholar] [CrossRef]
- Porter, N.J.; Christianson, N.H.; Decroos, C.; Christianson, D.W. Structural and Functional Influence of the Glycine-Rich Loop G 302 GGGY on the Catalytic Tyrosine of Histone Deacetylase 8. Biochemistry 2016, 55, 6718–6729. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucia-Campos, C.; Valenzuela, I.; Latorre-Pellicer, A.; Ros-Pardo, D.; Gil-Salvador, M.; Arnedo, M.; Puisac, B.; Castells, N.; Plaja, A.; Tenes, A.; et al. A Novel Intragenic Duplication in the HDAC8 Gene Underlying a Case of Cornelia de Lange Syndrome. Genes 2022, 13, 1413. https://doi.org/10.3390/genes13081413
Lucia-Campos C, Valenzuela I, Latorre-Pellicer A, Ros-Pardo D, Gil-Salvador M, Arnedo M, Puisac B, Castells N, Plaja A, Tenes A, et al. A Novel Intragenic Duplication in the HDAC8 Gene Underlying a Case of Cornelia de Lange Syndrome. Genes. 2022; 13(8):1413. https://doi.org/10.3390/genes13081413
Chicago/Turabian StyleLucia-Campos, Cristina, Irene Valenzuela, Ana Latorre-Pellicer, David Ros-Pardo, Marta Gil-Salvador, María Arnedo, Beatriz Puisac, Neus Castells, Alberto Plaja, Anna Tenes, and et al. 2022. "A Novel Intragenic Duplication in the HDAC8 Gene Underlying a Case of Cornelia de Lange Syndrome" Genes 13, no. 8: 1413. https://doi.org/10.3390/genes13081413
APA StyleLucia-Campos, C., Valenzuela, I., Latorre-Pellicer, A., Ros-Pardo, D., Gil-Salvador, M., Arnedo, M., Puisac, B., Castells, N., Plaja, A., Tenes, A., Cuscó, I., Trujillano, L., Ramos, F. J., Tizzano, E. F., Gómez-Puertas, P., & Pié, J. (2022). A Novel Intragenic Duplication in the HDAC8 Gene Underlying a Case of Cornelia de Lange Syndrome. Genes, 13(8), 1413. https://doi.org/10.3390/genes13081413