
An Integrative Genomic Prediction Approach for Predicting Buffalo Milk Traits by Incorporating Related Cattle QTLs

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Statistical Model
2.2. Animal Resources and Genomic Information
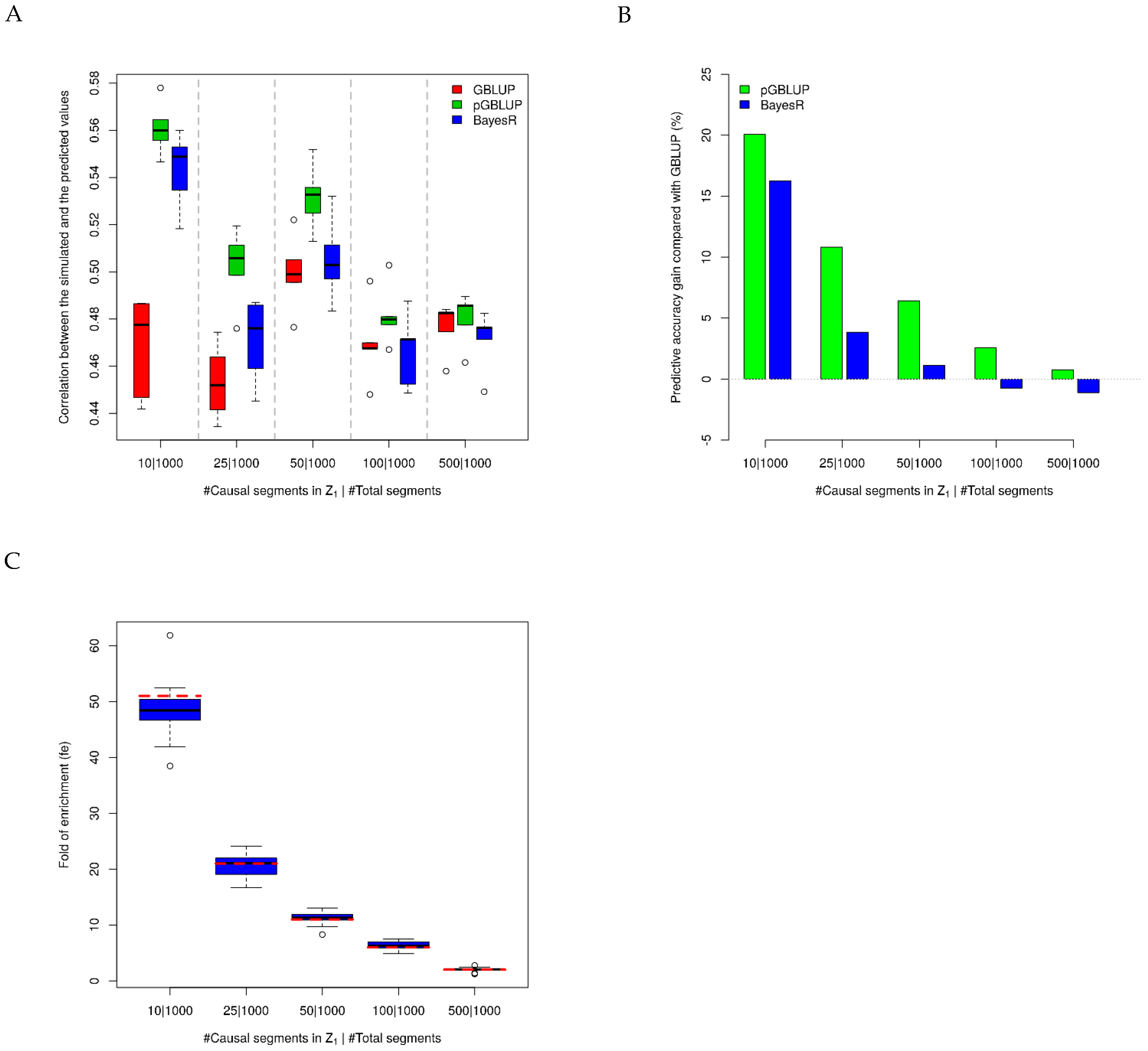
2.3. Simulations
- (1)
- Set the causal segments: The genotype matrix was standardized, and the 42,551 SNPs were divided into 1000 approximately equally sized segments, with 42 or 43 SNPs in each segment; s (10/25/50/100/500) segments were randomly selected as causal segments in our simulation settings, and the 10 SNPs in the center of each segment were then selected as causal SNPs; thus, the total number of causal SNPs (k) was 100/250/500/1000/5000, while the total number of SNPs in the causal segments was .
- (2)
- Simulate the SNP effects and phenotype: Firstly, all SNPs were simulated with the small effects following a normal distribution ; the k causal SNPs were simulated with additional effects following a normal distribution Then, the residual errors were sampled from a normal distribution , so that the total heritability of the simulated trait was 0.5. Based on Equation (1), for each individual, the phenotype was obtained as the summation of small effects, large effects, and the residual error.
- (3)
- Five-fold cross-validation: The 5024 individuals were divided into five groups, with 1004 or 1005 individuals in each group. Each time, one group of individuals was set as the test dataset, while the rest of the groups of individuals were set as the training dataset (i.e., five-fold cross-validation). We applied the pGBLUP approach in two ways to predict the performance in the test dataset: only the SNPs in the causal segments were set in ; SNPs in both the causal segments and non-causal segments were selected in . We also applied the traditional GBLUP method [3] and the BayesR method [16] to compare the performance. The GBLUP method assumes the effect size for every variant is sampled from the same normal distribution; the BayesR method uses an MCMC algorithm to estimate variant effects, which are modelled as a mixture distribution of four normal distributions, including a null distribution, , and three others: , , and , where is the additive genetic variance for the trait.
2.4. Genomic Prediction of Buffalo Milk Traits
2.5. Computation
3. Results
3.1. Predictive Accuracy in Simulations
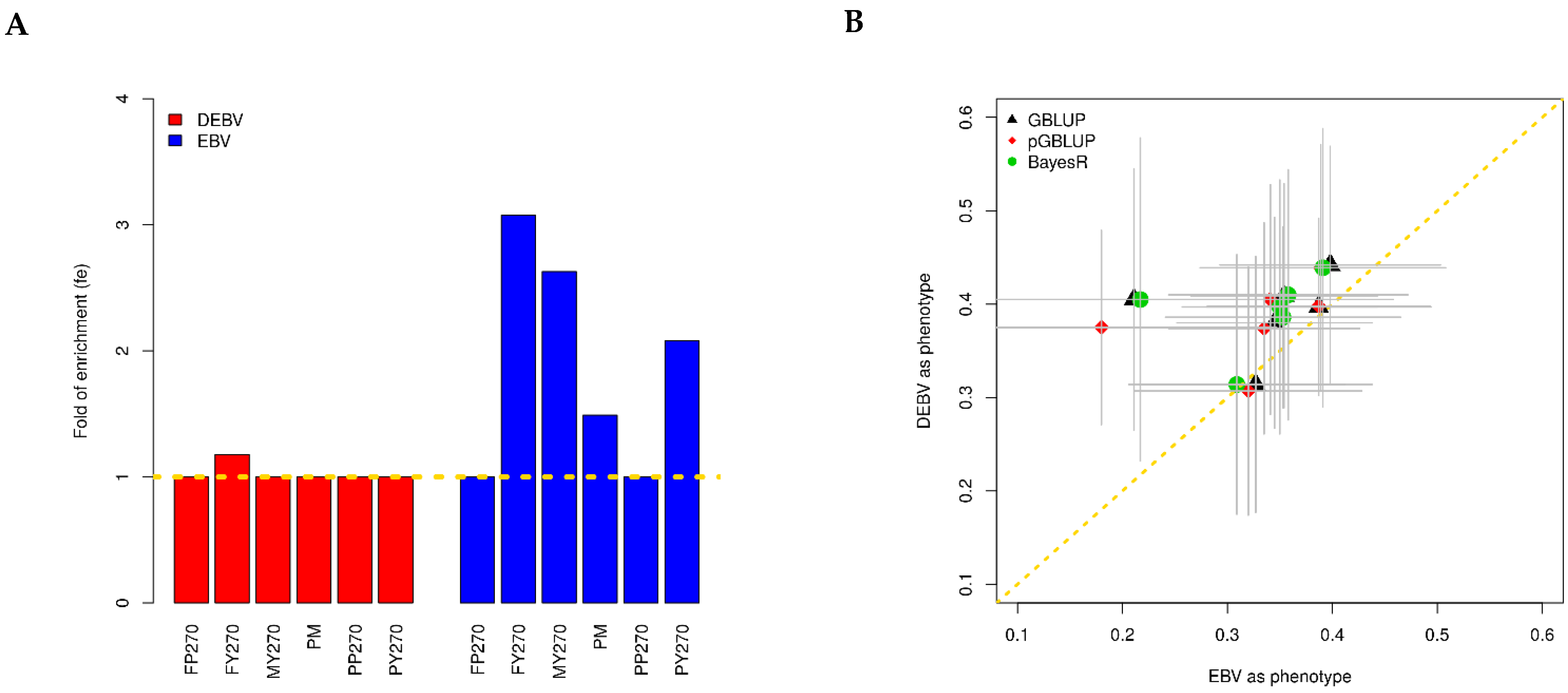
3.2. Genomic Prediction of Buffalo Milk Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| QTL | quantitative trait locus |
| QTN | quantitative trait nucleotide |
| SNP | single-nucleotide polymorphism |
| GBLUP | genomic best linear unbiased predictor |
| pGBLUP | incorporating prior biological information in genomic best linear unbiased predictor |
| EBV | estimated breeding value |
| GEBV | genomic estimated breeding value |
| DEBV | deregressed estimated breeding value |
| LMM | linear mixed models |
| fe | fold of enrichment |
References
- Hickey, J.M.; Chiurugwi, T.; Mackay, I.; Powell, W. Genomic prediction unifies animal and plant breeding programs to form platforms for biological discovery. Nat. Genet. 2017, 49, 1297. [Google Scholar] [CrossRef]
- Goddard, M.E.; Hayes, B.J. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 2009, 10, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Lee, S.H.; Weerasinghe, W.S.P.; Wray, N.R.; Goddard, M.E.; Van Der Werf, J.H. Using information of relatives in genomic prediction to apply effective stratified medicine. Sci. Rep. 2017, 7, 42091. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Shi, J.; García-Closas, M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 2016, 17, 392–406. [Google Scholar] [CrossRef]
- de los Campos, G.; Vazquez, A.I.; Fernando, R.; Klimentidis, Y.C.; Sorensen, D. Prediction of Complex Human Traits Using the Genomic Best Linear Unbiased Predictor. PLoS Genet. 2013, 9, e1003608. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Su, G.; Madsen, P. Partitioning additive genetic variance into genomic and remaining polygenic components for complex traits in dairy cattle. BMC Genet. 2012, 13, 44. [Google Scholar] [CrossRef]
- Coram, M.A.; Fang, H.; Candille, S.I.; Assimes, T.L.; Tang, H. Leveraging Multi-ethnic Evidence for Risk Assessment of Quantitative Traits in Minority Populations. Am. J. Hum. Genet. 2017, 101, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Speed, D.; Balding, D.J. MultiBLUP: Improved SNP-based prediction for complex traits. Genome Res. 2014, 24, 1550–1557. [Google Scholar] [CrossRef]
- Da, Y.; Wang, C.; Wang, S.; Hu, G. Mixed model methods for genomic prediction and variance component estimation of additive and dominance effects using SNP markers. PLoS ONE 2014, 9, e87666. [Google Scholar] [CrossRef]
- Kemper, K.E.; Reich, C.M.; Bowman, P.J.; Vander Jagt, C.J.; Chamberlain, A.J.; Mason, B.A.; Hayes, B.J.; Goddard, M.E. Improved precision of QTL mapping using a nonlinear Bayesian method in a multi-breed population leads to greater accuracy of across-breed genomic predictions. Genet. Sel. Evol. 2015, 47, 29. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef]
- Moser, G.; Lee, S.H.; Hayes, B.J.; Goddard, M.E.; Wray, N.R.; Visscher, P.M. Simultaneous discovery, estimation and prediction analysis of complex traits using a Bayesian mixture model. PLoS Genet. 2015, 11, e1004969. [Google Scholar] [CrossRef]
- Erbe, M.; Hayes, B.; Matukumalli, L.K.; Goswami, S.; Bowman, P.J.; Reich, C.M. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy Sci. 2012, 95, 4114–4129. [Google Scholar] [CrossRef]
- Zhou, X.; Carbonetto, P.; Stephens, M. Polygenic modeling with bayesian sparse linear mixed models. PLoS Genet. 2013, 9, e1003264. [Google Scholar] [CrossRef]
- Zeng, P.; Zhou, X. Non-parametric genetic prediction of complex traits with latent Dirichlet process regression models. Nat. Commun. 2017, 8, 456. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Zhou, X.; Yuan, X.; Zhao, S.; Li, X.; Liu, X. KAML: Improving genomic prediction accuracy of complex traits using machine learning determined parameters. Genome Biol. 2020, 21, 146. [Google Scholar] [CrossRef]
- MacLeod, I.; Bowman, P.; Vander Jagt, C.; Haile-Mariam, M.; Kemper, K.; Chamberlain, A.; Schrooten, C.; Hayes, B.; Goddard, M. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 144. [Google Scholar] [CrossRef]
- Gao, N.; Martini, J.W.R.; Zhang, Z.; Yuan, X.; Zhang, H.; Simianer, H.; Li, J. Incorporating Gene Annotation into Genomic Prediction of Complex Phenotypes. Genetics 2017, 207, 489–501. [Google Scholar] [CrossRef]
- Zhang, Z.; Erbe, M.; He, J.; Ober, U.; Gao, N.; Zhang, H.; Simianer, H.; Li, J. Accuracy of Whole-Genome Prediction Using a Genetic Architecture-Enhanced Variance-Covariance Matrix. G3 Genes Genomes Genet. 2015, 5, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Sahana, G.; Ma, P.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.S.; Sørensen, P. Exploring the genetic architecture and improving genomic prediction accuracy for mastitis and milk production traits in dairy cattle by mapping variants to hepatic transcriptomic regions responsive to intra-mammary infection. Genet. Sel. Evol. 2017, 49, 44. [Google Scholar] [CrossRef]
- Fragomeni, B.O.; Lourenco, D.A.L.; Masuda, Y.; Legarra, A.; Misztal, I. Incorporation of causative quantitative trait nucleotides in single-step GBLUP. Genet. Sel. Evol. 2017, 49, 59. [Google Scholar] [CrossRef] [PubMed]
- El-Halawany, N.; Abdel-Shafy, H.; Shawky, A.-E.-M.A.; Abdel-Latif, M.A.; Al-Tohamy, A.F.M.; Abd El-Moneim, O.M. Genome-wide association study for milk production in Egyptian buffalo. Livest. Sci. 2017, 198 (Suppl. C), 10–16. [Google Scholar] [CrossRef]
- Michelizzi, V.N.; Wu, X.; Dodson, M.V.; Michal, J.J.; Zambrano-Varon, J.; McLean, D.J.; Jiang, Z. A Global View of 54,001 Single Nucleotide Polymorphisms (SNPs) on the Illumina BovineSNP50 BeadChip and Their Transferability to Water Buffalo. Int. J. Biol. Sci. 2011, 7, 18–27. [Google Scholar] [CrossRef]
- Iamartino, D.; Williams, J.L.; Sonstegard, T.; Reecy, J.; Tassell Cv Nicolazzi, E.L.; Biffani, S.; Biscarini, F.; Schroeder, S.; de Oliveira, D.A. The buffalo genome and the application of genomics in animal management and improvement. Buffalo Bull. 2013, 32, 151–158. [Google Scholar]
- Iamartino, D.; Nicolazzi, E.L.; Van Tassell, C.P.; Reecy, J.M.; Fritz-Waters, E.R.; Koltes, J.E.; Biffani, S.; Sonstegard, T.S.; Schroeder, S.G.; Ajmone-Marsan, P. Design and validation of a 90K SNP genotyping assay for the water buffalo (Bubalus bubalis). PLoS ONE 2017, 12, e0185220. [Google Scholar] [CrossRef] [PubMed]
- De Camargo, G.; Aspilcueta-Borquis, R.R.; Fortes, M.; Porto-Neto, R.; Cardoso, D.F.; Santos, D.; Lehnert, S.; Reverter, A.; Moore, S.; Tonhati, H. Prospecting major genes in dairy buffaloes. BMC Genomics 2015, 16, 872. [Google Scholar] [CrossRef] [PubMed]
- Aspilcueta-Borquis, R.; Neto, F.A.; Santos, D.; Hurtado-Lugo, N.; Silva, J.; Tonhati, H. Multiple-trait genomic evaluation for milk yield and milk quality traits using genomic and phenotypic data in buffalo in Brazil. Gen. Mol. Res. 2015, 14, 18009–18017. [Google Scholar] [CrossRef] [PubMed]
- Borquis, R.R.A.; de Araujo Neto, F.R.; Baldi, F.; Hurtado-Lugo, N.; de Camargo, G.M.; Muñoz-Berrocal, M.; Tonhati, H. Multiple-trait random regression models for the estimation of genetic parameters for milk, fat, and protein yield in buffaloes. J. Dairy Sci. 2013, 96, 5923–5932. [Google Scholar] [CrossRef]
- Hossein-Zadeh, N.G.; Nazari, M.A.; Shadparvar, A.A. Genetic perspective of milk yield persistency in the first three lactations of Iranian buffaloes (Bubalus bubalis). J. Dairy Res. 2017, 84, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Patil, H.R. Genetic Evaluation of Fertility and Production Efficiency Traits in Murrah Buffalo; LUVAS: Hisar, India, 2016. [Google Scholar]
- Agudelo-Gómez, D.; Pelicioni Savegnago, R.; Buzanskas, M.; Ferraudo, A.; Prado Munari, D.; Cerón-Muñoz, M. Genetic principal components for reproductive and productive traits in dual-purpose buffaloes in Colombia. J. Anim. Sci. 2015, 93, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Chakravarty, A.; Singh, A.; Shivahre, P.R.; Upadhyay, A.; Sah, V.; Singh, K.M. Assessment of expected breeding values for fertility traits of Murrah buffaloes under subtropical climate. Vet. World 2015, 8, 320. [Google Scholar] [CrossRef]
- Gupta, J.P.; Sachdeva, G.K.; Gandhi, R.; Chakaravarty, A. Developing multiple-trait prediction models using growth and production traits in Murrah buffalo. Buffalo Bull. 2015, 34, 347–355. [Google Scholar]
- Aspilcueta-Borquis, R.; Neto, F.A.; Baldi, F.; Bignardi, A.; Albuquerque, L.G.; Tonhati, H. Genetic parameters for buffalo milk yield and milk quality traits using Bayesian inference. J. Dairy Sci. 2010, 93, 2195–2201. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Developmental progress and current status of the Animal QTLdb. Nucleic Acids Res. 2016, 44, D827–D833. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Edwards, S.M.; Thomsen, B.; Madsen, P.; Sorensen, P. Partitioning of genomic variance reveals biological pathways associated with udder health and milk production traits in dairy cattle. Genet. Sel. Evol. 2015, 47, 60. [Google Scholar] [CrossRef]
- Gusev, A.; Lee, S.H.; Trynka, G.; Finucane, H.; Vilhjálmsson, B.J.; Xu, H. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet. 2014, 95, 535–552. [Google Scholar] [CrossRef]
- Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.R.; Anttila, V. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015, 47, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X. A Unified Framework for Variance Component Estimation with Summary Statistics in Genome-wide Association Studies. Ann Appl Stat. 2017, 11, 2027–2051. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Sahana, G.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.S.; Sørensen, P. Integrating Sequence-based GWAS and RNA-Seq Provides Novel Insights into the Genetic Basis of Mastitis and Milk Production in Dairy Cattle. Sci. Rep. 2017, 7, 45560. [Google Scholar] [CrossRef] [PubMed]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef]
- Liu, J.J.; Liang, A.X.; Campanile, G.; Plastow, G.; Zhang, C.; Wang, Z.; Salzano, A.; Gasparrini, B.; Cassandro, M.; Yang, L.G. Genome-wide association studies to identify quantitative trait loci affecting milk production traits in water buffalo. J. Dairy Sci. 2018, 101, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Baldi, F.; Laureano, M.M.M.; Gordo, D.G.M.; Bignardi, A.B.; Borquis, R.R.A.; Albuquerque, L.G.; Tonhati, H. Effect of lactation length adjustment procedures on genetic parameter estimates for buffalo milk yield. Genet. Mol. Biol. 2011, 34, 62–67. [Google Scholar] [CrossRef]
- Gilmour, A.R.; Gogel, R.B.J.; Cullis, B.R.; Thompson, R. Asreml User Guide Release 3.0; 2009. Available online: https://asreml.kb.vsni.co.uk/wp-content/uploads/sites/3/2018/02/ASReml-3-User-Guide.pdf (accessed on 10 October 2017).
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A language for data analysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Zhang, Z.; Ober, U.; Erbe, M.; Zhang, H.; Gao, N.; He, J.; Li, J.; Simianer, H. Improving the accuracy of whole genome prediction for complex traits using the results of genome wide association studies. PLoS ONE 2014, 9, e93017. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Ding, X.; Bijma, P.; de Koning, D.J.; Zhang, Q. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS ONE 2010, 5, e12648. [Google Scholar] [CrossRef]
- Rohde, P.D.; Demontis, D.; Cuyabano, B.C.D.; Børglum, A.D.; Sørensen, P. Covariance association test (CVAT) identify genetic markers associated with schizophrenia in functionally associated biological processes. Genetics 2016, 203, 1901–1913. [Google Scholar] [CrossRef]
- Fang, L.; Sahana, G.; Ma, P.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.S.; Sørensen, P. Use of biological priors enhances understanding of genetic architecture and genomic prediction of complex traits within and between dairy cattle breeds. BMC Genomics 2017, 18, 604. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Pang, C.; Ma, X.; Duan, A.; Liang, S.; Lu, X.; Liang, X. Buffalo SREBP1: Molecular cloning, expression and association analysis with milk production traits. Anim. Genet. 2017, 48, 720–721. [Google Scholar] [CrossRef] [PubMed]
- Dinesh, K.; Verma, A.; Gupta, I.D.; Thakur, Y.P.; Verma, N.; Arya, A. Identification of polymorphism in exons 7 and 12 of lactoferrin gene and its association with incidence of clinical mastitis in Murrah buffalo. Trop. Anim. Health Prod. 2015, 47, 643–647. [Google Scholar] [CrossRef]
- El-Magd, M.A.; Abo-Al-Ela, H.G.; El-Nahas, A.; Saleh, A.A.; Mansour, A.A. Effects of a novel SNP of IGF2R gene on growth traits and expression rate of IGF2R and IGF2 genes in gluteus medius muscle of Egyptian buffalo. Gene 2014, 540, 133–139. [Google Scholar] [CrossRef]
- Yuan, J.; Zhou, J.; Deng, X.; Hu, X.; Li, N. Molecular cloning and single nucleotide polymorphism detection of buffalo DGAT1 gene. Biochem. Genet. 2007, 45, 611–621. [Google Scholar] [CrossRef]
- Rosati, A.; Van Vleck, L.D. Estimation of genetic parameters for milk, fat, protein and mozzarella cheese production for the Italian river buffalo Bubalus bubalis population. Livest. Prod. Sci. 2002, 74, 185–190. [Google Scholar] [CrossRef]
- Malhado, C.H.M.; Malhado, A.C.M.; Ramos, A.d.A.; Carneiro, P.L.S.; Souza JCd Pala, A. Genetic parameters for milk yield, lactation length and calving intervals of Murrah buffaloes from Brazil. Rev. Bras. De Zootec. 2013, 42, 565–569. [Google Scholar] [CrossRef]
- Ostersen, T.; Christensen, O.F.; Henryon, M.; Nielsen, B.; Su, G.; Madsen, P. Deregressed EBV as the response variable yield more reliable genomic predictions than traditional EBV in pure-bred pigs. Genet. Sel. Evol. 2011, 43, 38. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Trait | h2 | GBLUP | pGBLUP | BayesR | |
|---|---|---|---|---|---|
| DEBV | FP270 | 0.713 ± 0.112 | 0.314 ± 0.137 | 0.307 ± 0.133 | 0.314 ± 0.139 |
| FY270 | 0.703 ± 0.119 | 0.38 ± 0.113 | 0.374 ± 0.113 | 0.397 ± 0.136 | |
| MY270 | 0.753 ± 0.112 | 0.409 ± 0.12 | 0.405 ± 0.123 | 0.41 ± 0.134 | |
| PM | 0.702 ± 0.115 | 0.405 ± 0.14 | 0.375 ± 0.104 | 0.405 ± 0.173 | |
| PP270 | 0.75 ± 0.112 | 0.397 ± 0.095 | 0.398 ± 0.092 | 0.386 ± 0.097 | |
| PY270 | 0.793 ± 0.108 | 0.442 ± 0.127 | 0.439 ± 0.132 | 0.439 ± 0.149 | |
| EBV | FP270 | 0.741 ± 0.114 | 0.327 ± 0.111 | 0.32 ± 0.108 | 0.309 ± 0.103 |
| FY270 | 0.631 ± 0.124 | 0.345 ± 0.093 | 0.335 ± 0.091 | 0.35 ± 0.093 | |
| MY270 | 0.658 ± 0.122 | 0.354 ± 0.089 | 0.341 ± 0.077 | 0.358 ± 0.114 | |
| PM | 0.599 ± 0.123 | 0.211 ± 0.22 | 0.18 ± 0.167 | 0.217 ± 0.241 | |
| PP270 | 0.726 ± 0.115 | 0.387 ± 0.107 | 0.387 ± 0.106 | 0.353 ± 0.112 | |
| PY270 | 0.738 ± 0.116 | 0.398 ± 0.105 | 0.389 ± 0.101 | 0.391 ± 0.117 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, X.; Liang, A.; Plastow, G.; Zhang, C.; Wang, Z.; Liu, J.; Salzano, A.; Gasparrini, B.; Campanile, G.; Zhang, S.; et al. An Integrative Genomic Prediction Approach for Predicting Buffalo Milk Traits by Incorporating Related Cattle QTLs. Genes 2022, 13, 1430. https://doi.org/10.3390/genes13081430
Hao X, Liang A, Plastow G, Zhang C, Wang Z, Liu J, Salzano A, Gasparrini B, Campanile G, Zhang S, et al. An Integrative Genomic Prediction Approach for Predicting Buffalo Milk Traits by Incorporating Related Cattle QTLs. Genes. 2022; 13(8):1430. https://doi.org/10.3390/genes13081430
Chicago/Turabian StyleHao, Xingjie, Aixin Liang, Graham Plastow, Chunyan Zhang, Zhiquan Wang, Jiajia Liu, Angela Salzano, Bianca Gasparrini, Giuseppe Campanile, Shujun Zhang, and et al. 2022. "An Integrative Genomic Prediction Approach for Predicting Buffalo Milk Traits by Incorporating Related Cattle QTLs" Genes 13, no. 8: 1430. https://doi.org/10.3390/genes13081430
APA StyleHao, X., Liang, A., Plastow, G., Zhang, C., Wang, Z., Liu, J., Salzano, A., Gasparrini, B., Campanile, G., Zhang, S., & Yang, L. (2022). An Integrative Genomic Prediction Approach for Predicting Buffalo Milk Traits by Incorporating Related Cattle QTLs. Genes, 13(8), 1430. https://doi.org/10.3390/genes13081430