
Eye and Hair Color Prediction of Ancient and Second World War Skeletal Remains Using a Forensic PCR-MPS Approach

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bone Samples
2.2. DNA Extraction
2.3. DNA Quantification
2.4. HIrisPlex Sequencing on Ion S5
2.5. Sequencing Data Analysis and Genotyping
2.6. Generation of Consensus Genotype and Eye/Hair Color Prediction
2.7. STR-CE Typing
2.8. Elimination Database
2.9. Calculations and Graphs
3. Results
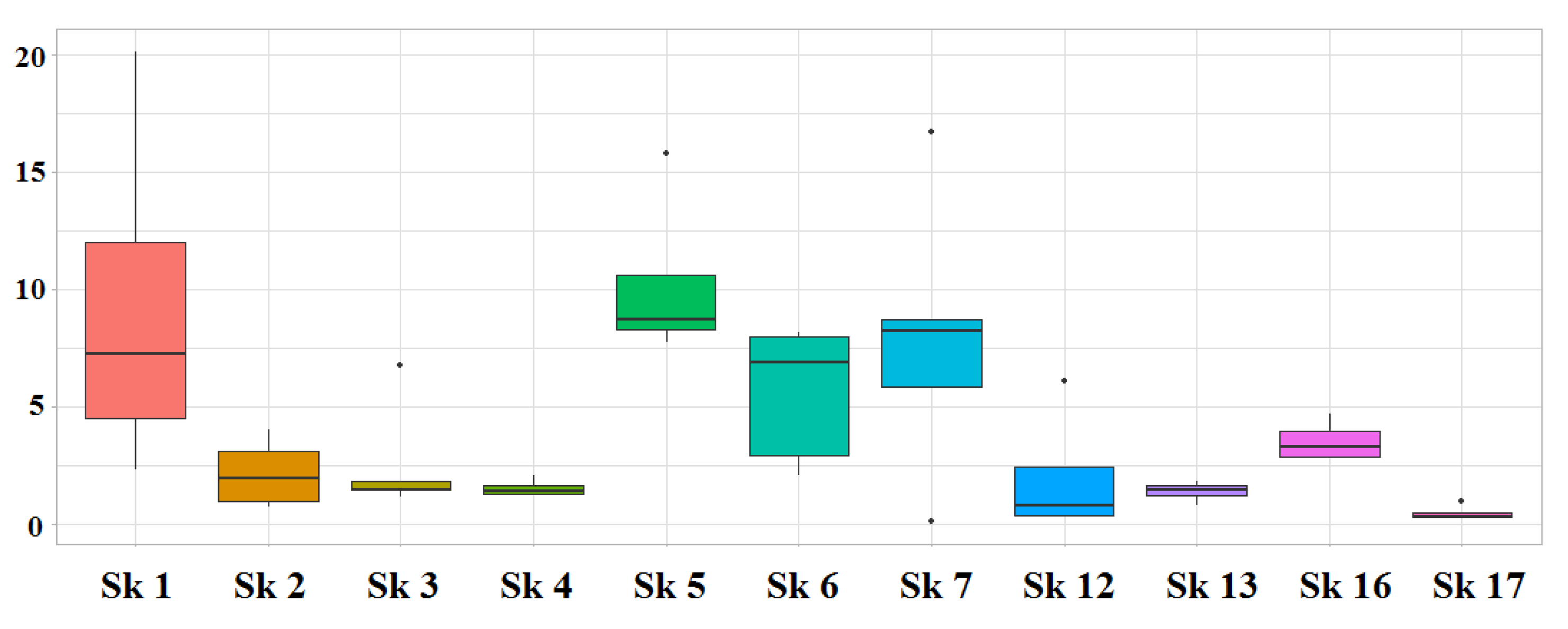
3.1. DNA Quantification
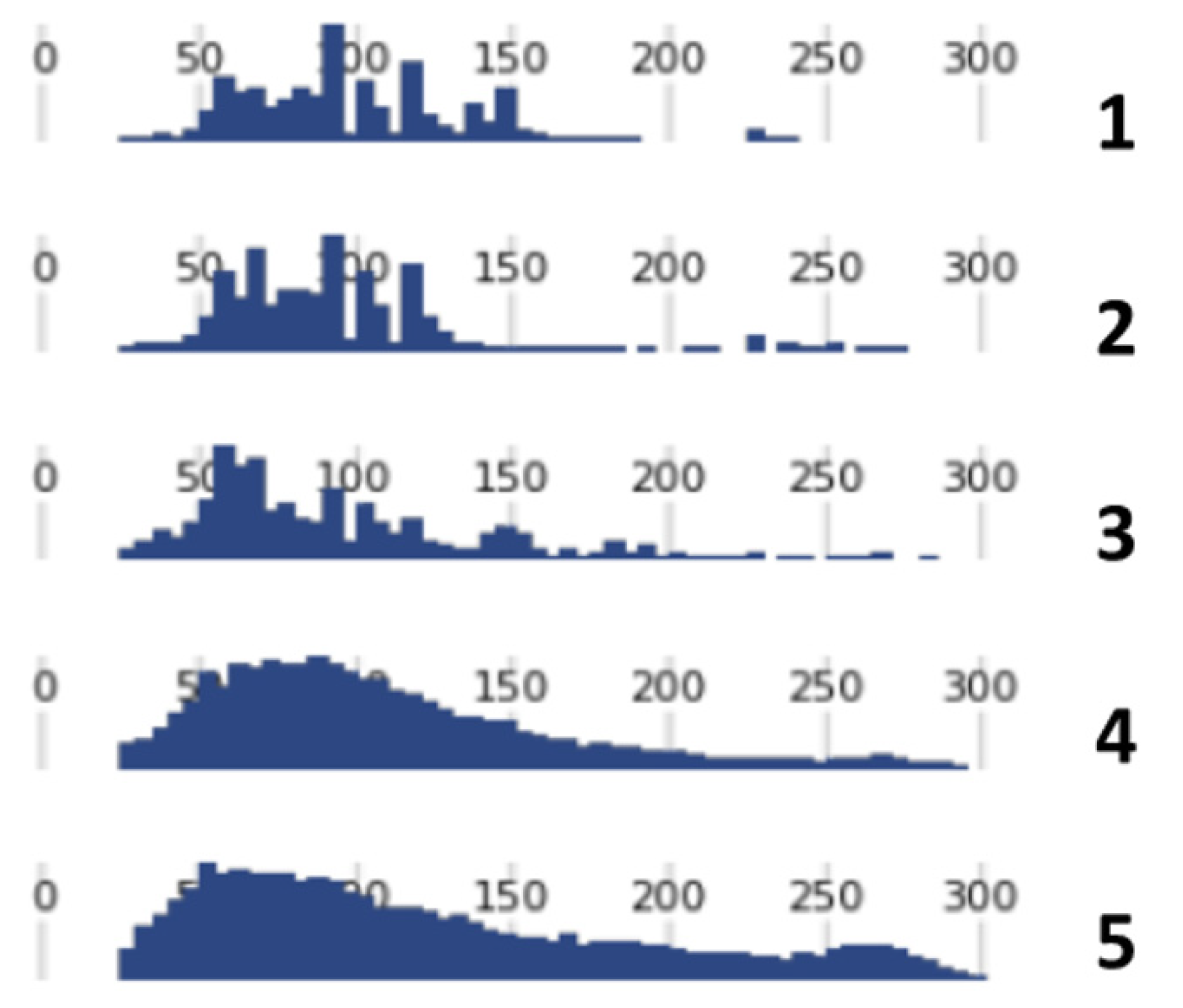
3.2. DNA Sequencing
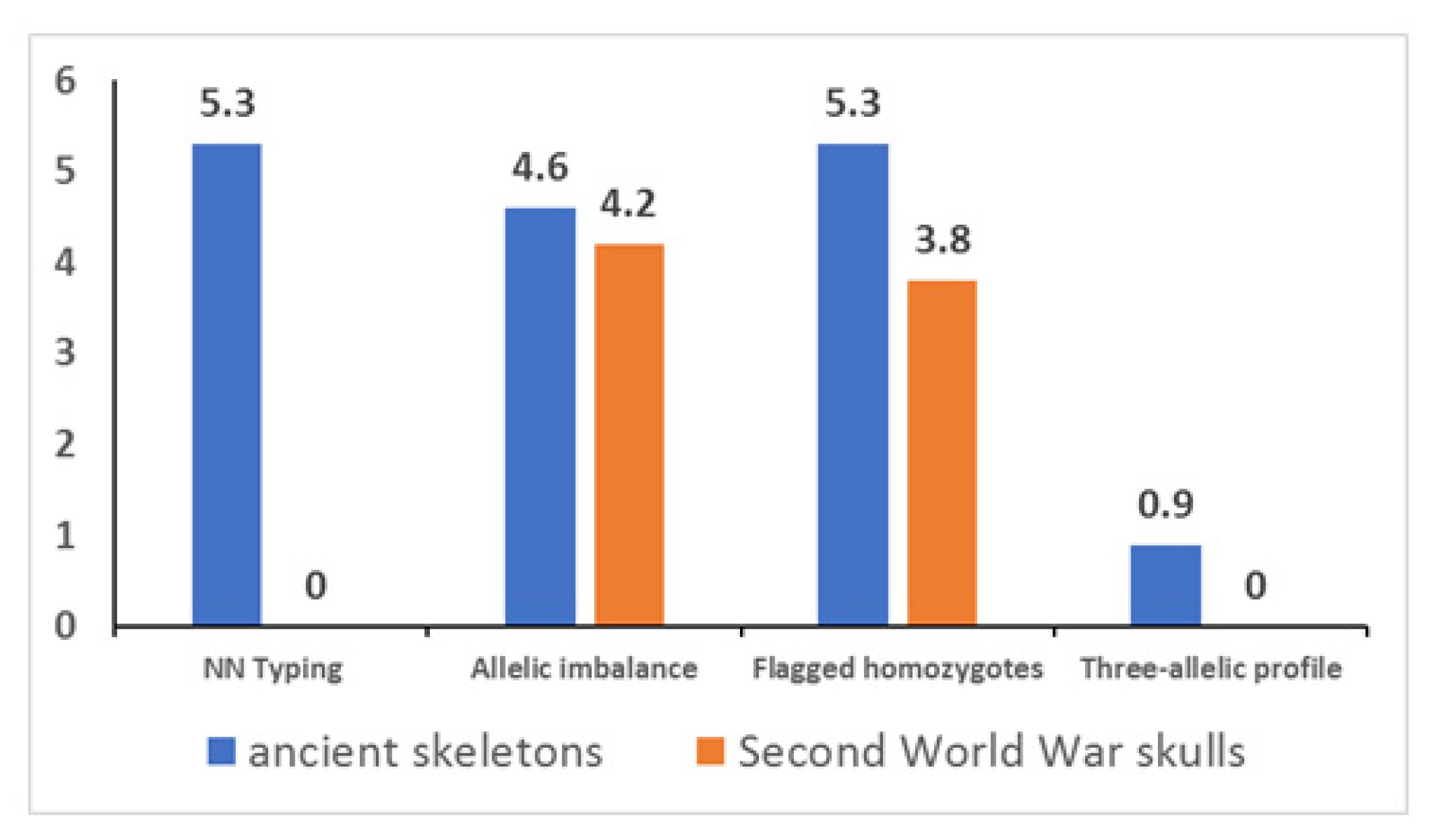
3.3. Genotyping
3.4. Consensus Genotype and Eye/Hair Color Prediction
3.5. Unmapped Reads and STR Typing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B


References
- McCord, B.R.; Gauthier, Q.; Cho, S.; Roig, M.N.; Gibson-Daw, G.C.; Young, B.; Taglia, F.; Zapico, S.C.; Mariot, R.F.; Lee, S.B.; et al. Forensic DNA Analysis. Anal. Chem. 2019, 91, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; van Duijn, K.; Vingerling, J.R.; Hofman, A.; Uitterlinden, A.G.; Janssens, A.C.; Kayser, M. Eye color and the prediction of complex phenotypes from genotypes. Curr. Biol. 2009, 19, R192–R193. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Lindenbergh, A.; Zuniga, S.B.; Sijen, T.; de Knijff, P.; Kayser, M.; Ballantyne, K.N. Developmental validation of the IrisPlex system: Determination of blue and brown iris colour for forensic intelligence. Forensic Sci. Int. Genet. 2011, 5, 464–471. [Google Scholar] [CrossRef]
- Walsh, S.; Liu, F.; Wollstein, A.; Kovatsi, L.; Ralf, A.; Kosiniak-Kamysz, A.; Branicki, W.; Kayser, M. The HIrisPlex system for simultaneous prediction of hair and eye colour from DNA. Forensic Sci. Int. Genet. 2013, 7, 98–115. [Google Scholar] [CrossRef]
- Branicki, W.; Liu, F.; van Duijn, K.; Draus-Barini, J.; Pośpiech, E.; Walsh, S.; Kupiec, T.; Wojas-Pelc, A.; Kayser, M. Model-based prediction of human hair color using DNA variants. Hum. Genet. 2011, 129, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Chaitanya, L.; Breslin, K.; Muralidharan, C.; Bronikowska, A.; Pospiech, E.; Koller, J.; Kovatsi, L.; Wollstein, A.; Branicki, W.; et al. Global skin colour prediction from DNA. Hum. Genet. 2017, 136, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Børsting, C.; Morling, N. Next generation sequencing and its applications in forensic genetics. Forensic Sci. Int. Genet. 2015, 18, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of nextgeneration sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Breslin, K.; Wills, B.; Ralf, A.; Ventayol Garcia, M.; Kukla-Bartoszek, M.; Pospiech, E.; Freire-Aradas, A.; Xavier, C.; Ingold, S.; de La Puente, M.; et al. HIrisPlex-S system for eye, hair, and skin color prediction from DNA: Massively parallel sequencing solutions for two common forensically used platforms. Forensic Sci. Int. Genet. 2019, 43, 102152. [Google Scholar] [CrossRef]
- Massively Parallel Sequencing Solutions for Human Identity. 2020. Available online: www.quigen.com (accessed on 10 January 2021).
- Verogen. ForenSeq DNA Signature Prep Kit. Available online: https://verogen.com/products/forenseq-dna-signature-prep-kit (accessed on 8 May 2022).
- Precision ID Ancestry Panel. Available online: https://www.thermofisher.com/order/catalog/product/A25642 (accessed on 6 June 2022).
- Xavier, C.; de la Puente, M.; Mosquera-Miguel, A.; Freire-Aradas, A.; Kalamara, V.; Vidaki, A.; Gross, T.E.; Revoir, A.; Pośpiech, E.; Kartasińska, E.; et al. Development and validation of the VISAGE AmpliSeq basic tool to predict appearance and ancestry from DNA. Forensic Sci. Int. Genet. 2020, 48, 102336. [Google Scholar] [CrossRef]
- Palencia-Madrid, L.; Xavier, C.; de la Puente, M.; Hohoff, C.; Phillips, C.; Kayser, M.; Parson, W. Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using PowerSeq Chemistry on the MiSeq FGx System. Genes 2020, 11, 708. [Google Scholar] [CrossRef] [PubMed]
- Bogdanowicz, W.; Allen, M.; Branicki, W.; Lembring, M.; Gajewska, M.; Kupiec, T. Genetic identification of putative remains of the famous astronomer Nicolaus Copernicus. Proc. Natl. Acad. Sci. USA 2009, 106, 12279–12282. [Google Scholar] [CrossRef] [PubMed]
- Draus-Barini, J.; Walsh, S.; Pośpiech, E.; Kupiec, T.; Głąb, H.; Branicki, W.; Kayser, M. Bona fide colour: DNA prediction of human eye and hair colour from ancient and contemporary skeletal remains. Investig. Genet. 2013, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Fortes, G.G.; Balaresque, P.; Thomas, M.G.; Balding, D.; Maisano Delser, P.; Neumann, R.; Parson, W.; Knapp, M.; Walsh, S.; et al. Identification of the remains of King Richard III. Nat. Commun. 2014, 5, 5631. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, M.; Haas, C.; Lösch, S.; Moghaddam, N.; Villa, I.M.; Walsh, S.; Kayser, M.; Seiler, R.; Ruehli, F.; Janosa, M.; et al. Multidisciplinary Identification of the Controversial Freedom Fighter Jörg Jenatsch, Assassinated 1639 in Chur, Switzerland. PLoS ONE 2016, 28, e0168014. [Google Scholar] [CrossRef]
- Peltzer, A.; Mittnik, A.; Wang, C.C.; Begg, T.; Posth, C.; Nieselt, K.; Krause, J. Inferring genetic origins and phenotypic traits of George Bähr, the architect of the Dresden Frauenkirche. Sci. Rep. 2018, 8, 2115. [Google Scholar] [CrossRef] [PubMed]
- Schablitsky, J.M.; Witt, K.E.; Madrigal, J.R.; Ellegaard, M.R.; Malhi, R.S.; Schroeder, H. Ancient DNA analysis of a nineteenth century tobacco pipe from a Maryland slave quarter. J. Archeol. Sci. 2019, 105, 11–18. [Google Scholar] [CrossRef]
- Schmidt, N.; Schücker, K.; Krause, I.; Dörk, T.; Klintschar, M.; Hummel, S. Genome-wide SNP typing of ancient DNA: Determination of hair and eye color of Bronze Age humans from their skeletal remains. Am. J. Phys. Anthropol. 2020, 172, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, L.; Pajnič, I.Z.; Walsh, S.; Balažic, J.; Zupanc, T.; Kayser, M. Bringing colour back after 70 years: Predicting eye and hair colour from skeletal remains of World War II victims using the HIrisPlex system. Forensic Sci. Int. Genet. 2017, 26, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Samida, S.; Feuchter, J. Why archaeologists, historians and geneticists should work together–and how. Mediev. Worlds 2016, 4, 5–21. [Google Scholar] [CrossRef]
- Capelli, C.; Tschentscher, F.; Pascali, V.L. “Ancient” protocols for the crime scene? Similarities and differences between forensic genetics and ancient DNA analysis. Forensic Sci. Int. 2003, 131, 59–64. [Google Scholar] [CrossRef]
- Hagelberg, E.; Hofreiter, M.; Keyser, C. Introduction. Ancient DNA: The first three decades. Philos. Trans. R Soc. Lond. B Biol. Sci. 2015, 370, 20130371. [Google Scholar] [CrossRef]
- Orlando, L.; Allaby, R.; Skoglund, P.; Der Sarkissian, C.; Stockhammer, P.W.; Avila, M.; Fu, Q.; Krause, J.; Willerslev, E.; Stone, A.; et al. Ancient DNA analysis. Nat. Rev. Methods Primers 2021, 1, 14. [Google Scholar] [CrossRef]
- Kukla-Bartoszek, M.; Szargut, M.; Pośpiech, E.; Diepenbroek, M.; Zielińska, G.; Jarosz, A.; Piniewska-Róg, D.; Arciszewska, J.; Cytacka, S.; Spólnicka, M.; et al. The challenge of predicting human pigmentation traits in degraded bone samples with the MPS-based HIrisPlex-S system. Forensic Sci. Int. Genet. 2020, 47, 102301. [Google Scholar] [CrossRef]
- Hernandez, C.J.; Majeska, R.J.; Schaffler, M.B. Osteocyte density in woven bone. Bone 2004, 35, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Pinhasi, R.; Fernandes, D.; Sirak, K.; Novak, M.; Connell, S.; Alpaslan-Roodenberg, S.; Gerritsen, F.; Moiseyev, V.; Gromov, A.; Raczky, P.; et al. Optimal Ancient DNA Yields from the Inner Ear Part of the Human Petrous Bone. PLoS ONE 2015, 10, e0129102. [Google Scholar] [CrossRef] [PubMed]
- Pilli, E.; Vai, S.; Caruso, M.G.; D’Errico, G.; Berti, A.; Caramelli, D. Neither femur nor tooth: Petrous bone for identifying archaeological bone samples via forensic approach. Forensic Sci. Int. 2018, 283, 144–149. [Google Scholar] [CrossRef]
- Misner, L.M.; Halvorson, A.C.; Dreier, J.L.; Ubelaker, D.H.; Foran, D.R. The correlation between skeletal weathering and DNA quality and quantity. J. Forensic Sci. 2009, 54, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Cannet, C.; Zvénigorosky, V.; Geraut, A.; Koch, G.; Delabarde, T.; Ludes, B.; Raul, J.S.; Keyser, C. The petrous bone: Ideal substrate in legal medicine? Forensic Sci. Int. Genet. 2020, 47, 102305. [Google Scholar] [CrossRef]
- Gaudio, D.; Fernandes, D.M.; Schmidt, R.; Cheronet, O.; Mazzarelli, D.; Mattia, M.; O’Keeffe, T.; Feeney, R.N.M.; Cattaneo, C.; Pinhasi, R. Genome-Wide DNA from Degraded Petrous Bones and the Assessment of Sex and Probable Geographic Origins of Forensic Cases. Sci. Rep. 2019, 3, 8226. [Google Scholar] [CrossRef]
- Kulstein, G.; Hadrys, T.; Wiegand, P. As solid as a rock-comparison of CE- and MPS-based analyses of the petrosal bone as a source of DNA for forensic identification of challenging cranial bones. Int. J. Legal Med. 2018, 132, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ferenc, M. Topografija evidentiranih grobišč. In Poročilo Komisije Vlade Republike Slovenije za Reševanje Vprašanj Prikritih Grobišč 2005–2008; Dežman, J., Ed.; Družina: Ljubljana, Slovenia, 2008; pp. 7–27. [Google Scholar]
- Poinar, H. The top 10 list: Criteria of authenticity for DNA from ancient and forensic samples. Int. Congr. Ser. 2003, 1239, 575–579. [Google Scholar] [CrossRef]
- Pääbo, S.; Poinar, H.; Serre, D.; Jaenicke-Despres, V.; Hebler, J.; Rohland, N.; Kuch, M.; Krause, J.; Vigilant, L.; Hofreiter, M. Genetic analyses from ancient DNA. Annu. Rev. Genet. 2004, 38, 645–679. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Hofreiter, M. Ancient DNA extraction from bones and teeth. Nat. Protoc. 2007, 2, 1756. [Google Scholar] [CrossRef]
- Llamas, B.; Valverde, G.; Fehren-Schmitz, L.; Weyrich, L.S.; Cooper, A.; Haak, W. From the field to the laboratory: Controlling DNA contamination in human ancient DNA research in the high-throughput sequencing era. Sci. Technol. Archaeol. Res. 2017, 1, 1–14. [Google Scholar] [CrossRef]
- Hofreiter, M.; Sneberger, J.; Pospisek, M.; Vanek, D. Progress in forensic bone DNA analysis: Lessons learned from ancient DNA. Forensic Sci. Int. Genet. 2021, 54, 102538. [Google Scholar] [CrossRef]
- Zupanič Pajnič, I. Extraction of DNA from human skeletal material. In Forensic DNA Typing Protocols; Goodwin, W., Ed.; Methods in Molecular Biology; Humana: New York, NY, USA, 2016; Volume 1420, pp. 89–108. [Google Scholar]
- Promega Corporation. PowerQuant System. Technical Manual; Promega Corporation: Madison, WI, USA, 2019. [Google Scholar]
- Thermo Fisher Scientific. Precision ID SNP Panel with the HID Ion S5TM/HID Ion Gene StudioTM S5 System, Application Guide, MAN0017767; Thermo Fisher Scientific: Carlsbad, CA, USA, 2019. [Google Scholar]
- Thermo Fisher Scientific. Torrent Suite™ Software 5.10, User Guide, MAN0017598; Thermo Fisher Scientific: Carlsbad, CA, USA, 2018. [Google Scholar]
- Samtools. Available online: http://www.htslib.org (accessed on 4 June 2020).
- Python. Available online: http://www.python.org (accessed on 4 June 2020).
- Babraham Bioinformatics. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 4 June 2020).
- National Library of Medicine. Basic Local Alignment Search Tool. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 16 May 2021).
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA damage. Cold Spring Harb Perspect Biol. 2013, 5, a012567. [Google Scholar] [CrossRef]
- Turchi, C.; Previderè, C.; Bini, C.; Carnevali, E.; Grignani, P.; Manfredi, A.; Melchionda, F.; Onofri, V.; Pelotti, S.; Robino, C.; et al. Assessment of the Precision ID Identity Panel kit on challenging forensic samples. Forensic Sci. Int. Genet. 2020, 49, 102400. [Google Scholar] [CrossRef]
- Bruijns, B.; Tiggelaar, R.; Gardeniers, H. Massively parallel sequencing techniques for forensics: A review. Electrophoresis 2018, 39, 2642–2654. [Google Scholar] [CrossRef]
- Salata, E.; Agostino, A.; Ciuna, I.; Wootton, S.; Ripani, L.; Berti, A. Revealing the challenges of low template DNA analysis with the prototype Ion AmpliSeqTM Identity panel v2.3 on the PGMTM Sequencer. Forensic Sci. Int. Genet. 2016, 22, 25–36. [Google Scholar] [CrossRef]
- Korlević, P.; Gerber, T.; Gansauge, M.T.; Hajdinjak, M.; Nagel, S.; Aximu-Petri, A.; Meyer, M. Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Biotechniques 2015, 59, 87–93. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. dbSNP. Available online: https://www.ncbi.nlm.nih.gov/snp (accessed on 20 April 2022).
- Fattorini, P.; Marrubini, G.; Ricci, U.; Gerin, F.; Grignani, P.; Cigliero, S.; Xamin, A.; Edalucci, E.; La Marca, G.; Previderé, C. Estimating the integrity of aged DNA samples by CE. Electrophoresis 2009, 30, 3986–3995. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Chaitanya, L.; Clarisse, L.; Wirken, L.; Draus-Barini, J.; Kovatsi, L.; Maeda, H.; Ishikawa, T.; Sijen, T.; de Knijff, P.; et al. Developmental validation of the HIrisPlex system: DNA-based eye and hair colour prediction for forensic and anthropological usage. Forensic Sci. Int. Genet. 2014, 9, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Bragg, L.M.; Stone, G.; Butler, M.K.; Hugenholtz, P.; Tyson, G.W. Shining a light on dark sequencing: Characterising errors in Ion Torrent PGM data. PLoS Comput. Biol. 2013, 9, e1003031. [Google Scholar] [CrossRef]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef]
- Ballard, D.; Winkler-Galicki, J.; Wesoły, J. Massive parallel sequencing in forensics: Advantages, issues, technicalities, and prospects. Int. J. Legal Med. 2020, 134, 1291–1303. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Skeleton | Sample | Marker | Genotype | % | ||
|---|---|---|---|---|---|---|
| Sk_1 | #141 | rs683 | A (2576) | C (3930) | T (707) | 9.8 |
| Sk_1 | #142 | rs1042602 | A (16538) | C (23040) | T (1079) | 2.7 |
| Sk_4 | #97 | rs12896399 | G (7075) | T (5543) | A (982) | 7.2 |
| Sk_4 | #96 | rs12896399 | G (12904) | T (6626) | A (432) | 2.2 |
| Sk_4 | #96 | rs16891982 | C (4217) | G (1943) | T (199) | 3.1 |
| Sk_6 | #59 | rs4959270 | A (1824) | C (5200) | T (236) | 3.3 |
| Sk_6 | #6D | rs683 | A (15494) | C (13807) | T (1455) | 4.7 |
| Sk_17 | #176 | rs1042602 | C (2943) | A (296) | T (707) | 17.9 |
| Sample | Dating | Libraries | Snp | Eye Color | Hair Color | ||
|---|---|---|---|---|---|---|---|
| Prediction | AUC Loss | Prediction | AUC Loss | ||||
| Sk_1 | 16th | 4 (0) | 24 | blue | 0 | brown/dark-brown | 0 |
| Sk_2 | 16th | 4 (1) | 23 | brown | 0 | dark-brown/black | 0.148 |
| Sk_3 | 16th | 5 (1) | 24 | brown | 0 | dark-brown/black | 0 |
| Sk_4 | 18th | 4 (0) | 22 | brown | 0 | dark-brown/black | 0.013 |
| Sk_5 | 17th | 4 (0) | 24 | blue | 0 | dark-blond/brown | 0 |
| Sk_6 | 18th | 5 (0) | 22 | und. | 0.727 | brown/dark-brown | 0.305 |
| Sk_7 | 18th | 6 (3) | 16 | blue | 0.066 | blond/dark-blond | 0.064 |
| Sk_12 | 3th | 4 (0) | 23 | brown | 0.043 | brown/dark-brown | 0 |
| Sk_13 | 3th | 4 (0) | 23 | brown | 0 | dark-brown/black | 0.003 |
| Sk_16 | 4th | 4 (3) | 21 * | brown | 0 | brown/dark-brown | 0.02 |
| Sk_17 | 4th | 4 (0) | 19 | und. | 0.812 | dark-blond/brown | 0.316 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zupanič Pajnič, I.; Zupanc, T.; Leskovar, T.; Črešnar, M.; Fattorini, P. Eye and Hair Color Prediction of Ancient and Second World War Skeletal Remains Using a Forensic PCR-MPS Approach. Genes 2022, 13, 1432. https://doi.org/10.3390/genes13081432
Zupanič Pajnič I, Zupanc T, Leskovar T, Črešnar M, Fattorini P. Eye and Hair Color Prediction of Ancient and Second World War Skeletal Remains Using a Forensic PCR-MPS Approach. Genes. 2022; 13(8):1432. https://doi.org/10.3390/genes13081432
Chicago/Turabian StyleZupanič Pajnič, Irena, Tomaž Zupanc, Tamara Leskovar, Matija Črešnar, and Paolo Fattorini. 2022. "Eye and Hair Color Prediction of Ancient and Second World War Skeletal Remains Using a Forensic PCR-MPS Approach" Genes 13, no. 8: 1432. https://doi.org/10.3390/genes13081432
APA StyleZupanič Pajnič, I., Zupanc, T., Leskovar, T., Črešnar, M., & Fattorini, P. (2022). Eye and Hair Color Prediction of Ancient and Second World War Skeletal Remains Using a Forensic PCR-MPS Approach. Genes, 13(8), 1432. https://doi.org/10.3390/genes13081432