
Molecular Dynamic Simulation Reveals Structure Differences in APOL1 Variants and Implication in Pathogenesis of Chronic Kidney Disease

 , and
, and
Abstract
:1. Introduction
2. Methodology
2.1. Protein and Ligand Preparation
2.2. Molecular Dynamic Simulation
3. Results
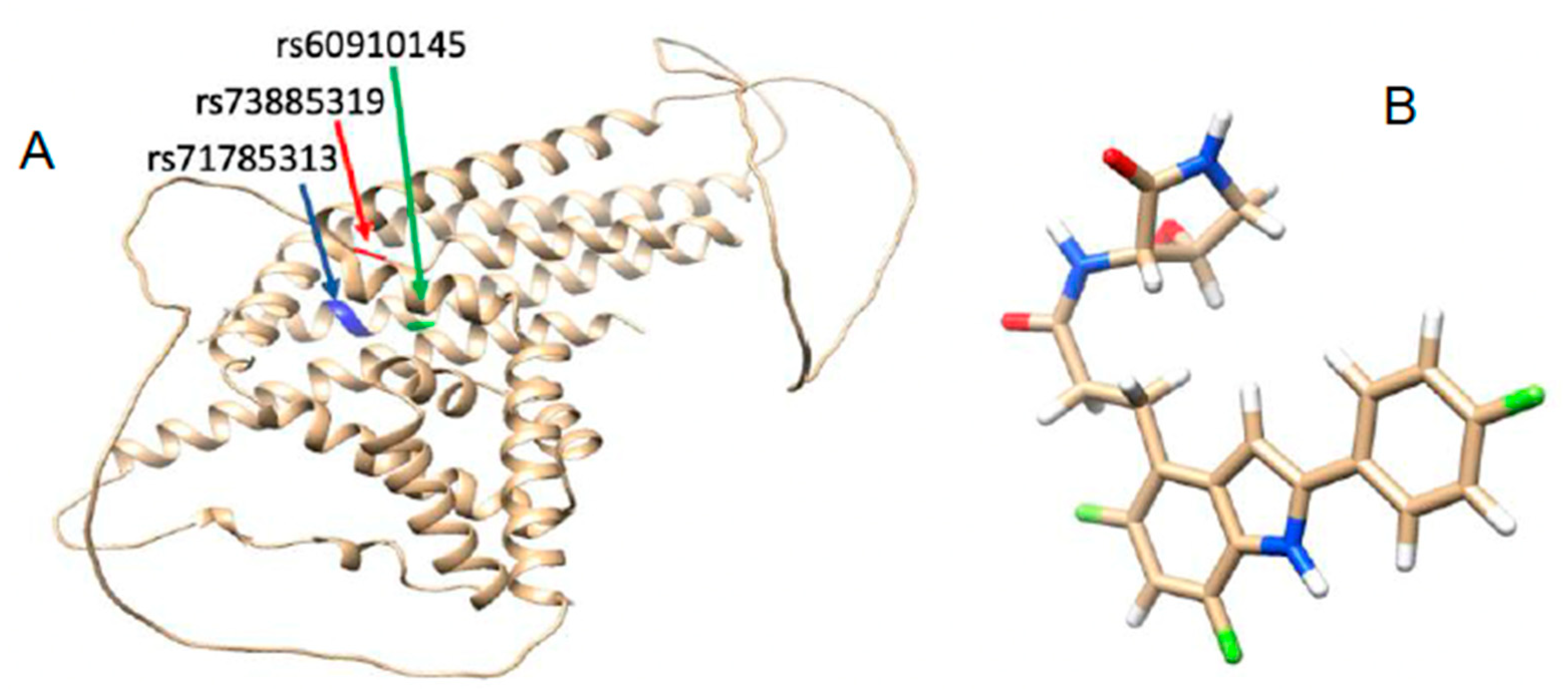
3.1. APOL1 Structural Elucidation
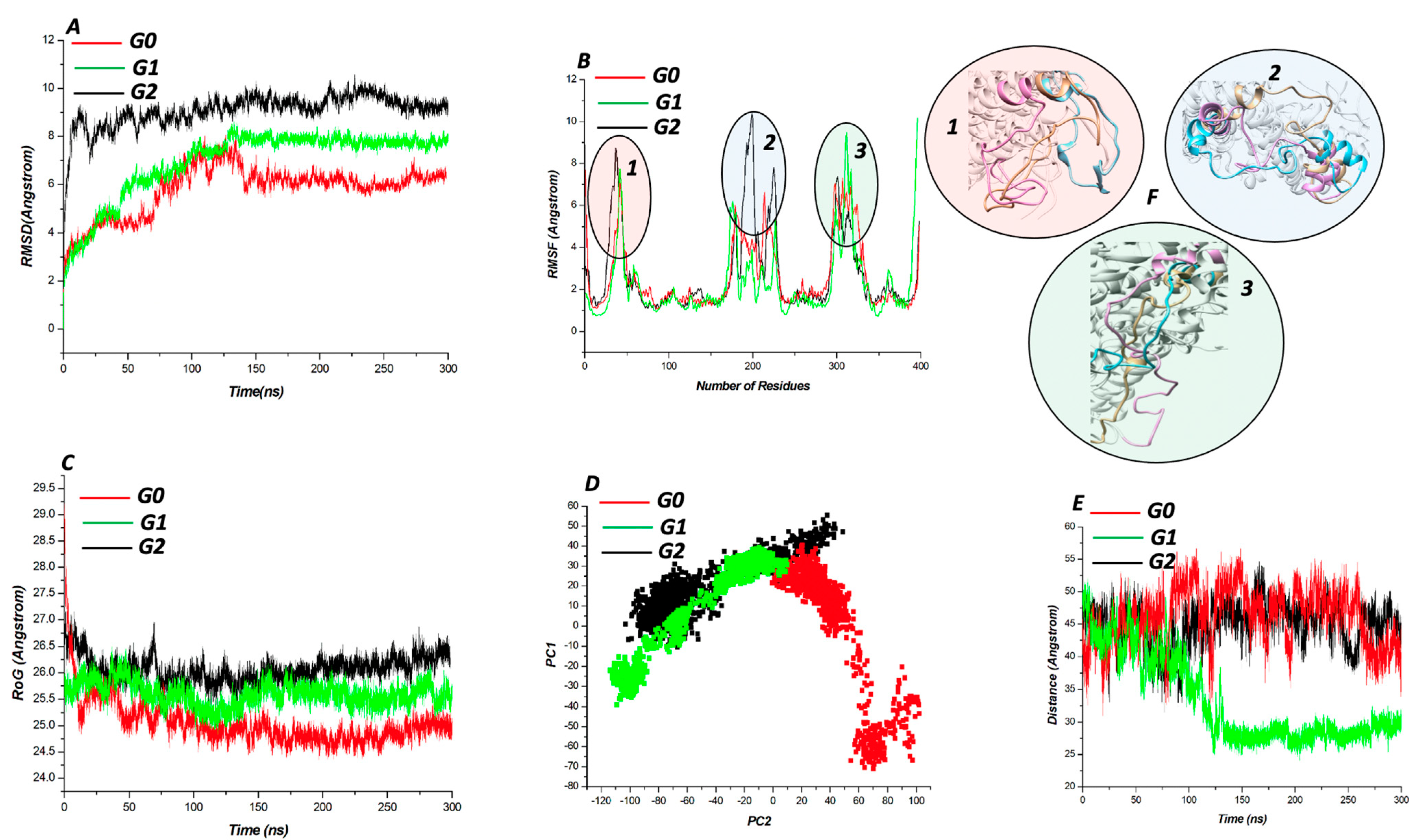
3.2. Mutation-Structural Perturbation of APOL1
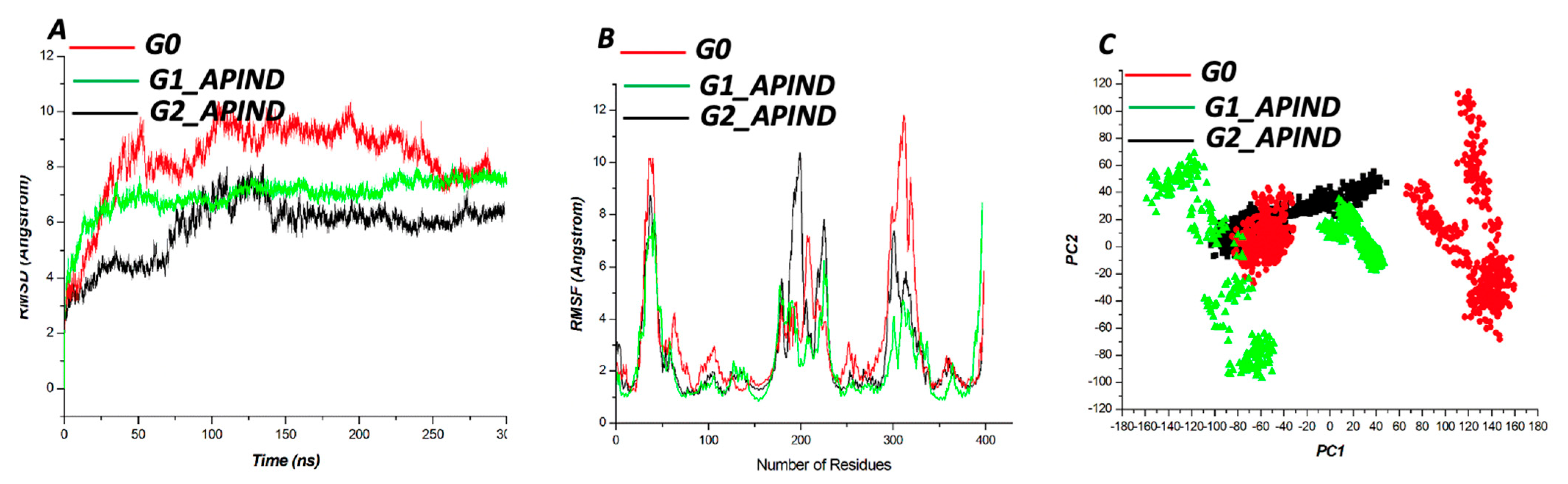
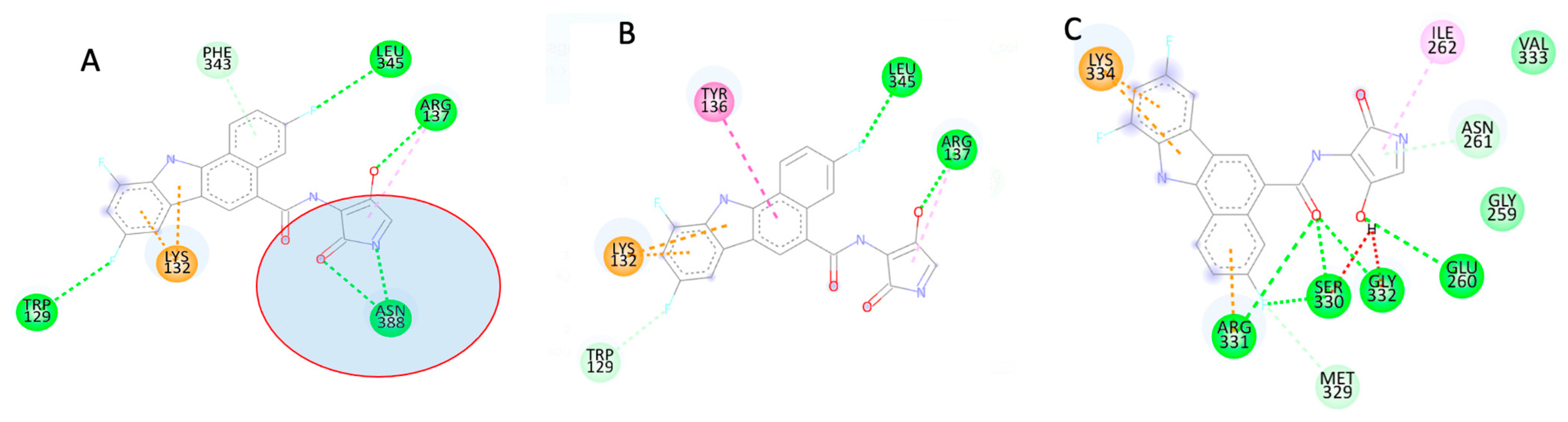
APIND Alters the Structure of Apol1
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lv, J.-C.; Zhang, L.-X. Prevalence and Disease Burden of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease Statistics for the United States|NIDDK. Available online: https://www.niddk.nih.gov/health-information/health-statistics/kidney-disease (accessed on 7 March 2022).
- Kidney Disease/Chronic Kidney Disease: Symptoms, Treatment & More. Available online: https://my.clevelandclinic.org/health/diseases/15096-kidney-disease-chronic-kidney-disease (accessed on 7 March 2022).
- Things to Know About Kidney Function|National Kidney Foundation. Available online: https://www.kidney.org/kidneydisease/howkidneyswrk (accessed on 7 March 2022).
- Kaze, A.D.; Ilori, T.; Jaar, B.G.; Echouffo-Tcheugui, J.B. Burden of chronic kidney disease on the African continent: A systematic review and meta-analysis. BMC Nephrol. 2018, 19, 125. [Google Scholar] [CrossRef] [PubMed]
- Chronic Kidney Disease (CKD)—Symptoms, Causes, Treatment|National Kidney Foundation. Available online: https://www.kidney.org/atoz/content/about-chronic-kidney-disease (accessed on 7 March 2022).
- Reidy, K.J.; Hjorten, R.; Parekh, R.S. Genetic risk of APOL1 and kidney disease in children and young adults of African ancestry. Curr. Opin. Pediatr. 2018, 30, 252. [Google Scholar] [CrossRef] [PubMed]
- Hung, R.K.; Binns-Roemer, E.; Booth, J.W.; Hilton, R.; Harber, M.; Santana-Suarez, B.; Campbell, L.; Fox, J.; Ustianowski, A.; Cosgrove, C.; et al. Genetic Variants of APOL1 Are Major Determinants of Kidney Failure in People of African Ancestry with HIV. Kidney Int. Rep. 2022, 7, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, L.A.; O’Toole, J.F.; Sedor, J.R. Identifying the Intracellular Function of APOL1. J. Am. Soc. Nephrol. 2017, 28, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Capewell, P.; Clucas, C.; Veitch, N.; Weir, W.; Thomson, R.; Raper, J.; MacLeod, A. A Primate APOL1 Variant That Kills Trypanosoma brucei gambiense. PLoS Negl. Trop. Dis. 2016, 10, e0004903. [Google Scholar] [CrossRef]
- Yusuf, A.A.; Govender, A.M.; Brandenburg, J.-T.; Winkler, A.C. Kidney disease and APOL1. Hum. Mol. Genet. 2021, 30, R129–R137. [Google Scholar] [CrossRef] [PubMed]
- Thurlow, J.S.; Joshi, M.; Yan, G.; Norris, K.C.; Agodoa, L.Y.; Yuan, C.M.; Nee, R. Global Epidemiology of End-Stage Kidney Disease and Disparities in Kidney Replacement Therapy. Am. J. Nephrol. 2021, 52, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Pays, E. The function of apolipoproteins L (APOLs): Relevance for kidney disease, neurotransmission disorders, cancer and viral infection. FEBS J. 2021, 288, 360–381. [Google Scholar] [CrossRef] [PubMed]
- Patient Education: Dialysis or Kidney Transplantation—Which Is Right for Me? (Beyond the Basics)–UpToDate. Available online: https://www.uptodate.com/contents/dialysis-or-kidney-transplantation-which-is-right-for-me-beyond-the-basics (accessed on 7 March 2022).
- Duriseti, P.; Idrees, N.; Aldairem, A.; Jaber, B.L.; Balakrishnan, V.S. Cost analysis of two modalities of continuous renal replacement therapy. Hemodial. Int. 2021, 25, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, R.W. Novel APOL1 Inhibitors for Treating Kidney Diseases. ACS Med. Chem. Lett. 2020, 11, 2352–2353. [Google Scholar] [CrossRef] [PubMed]
- AlphaFold Protein Structure Database. Available online: https://alphafold.ebi.ac.uk/ (accessed on 7 March 2022).
- Molegro Molecular Viewer—Molexus. Available online: http://molexus.io/molegro-molecular-viewer/ (accessed on 7 March 2022).
- Download UCSF Chimera. Available online: https://www.cgl.ucsf.edu/chimera/download.html (accessed on 7 March 2022).
- Chimera User’s Guide. Available online: https://www.cgl.ucsf.edu/chimera/current/docs/UsersGuide/ (accessed on 7 March 2022).
- Marvin|ChemAxon. Available online: https://chemaxon.com/products/marvin (accessed on 7 March 2022).
- Avogadro—Free Cross-Platform Molecular Editor–Avogadro. Available online: https://avogadro.cc/ (accessed on 7 March 2022).
- The Amber Force Fields. Available online: https://ambermd.org/AmberModels.php (accessed on 7 March 2022).
- Harrach, M.F.; Drossel, B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef] [PubMed]
- Snake Algorithm–File Exchange—MATLAB Central. Available online: https://www.mathworks.com/matlabcentral/fileexchange/51220-snake-algorithm (accessed on 7 March 2022).
- Okoh, M.P.; Singla, R.K.; Madu, C.; Soremekun, O.; Adejoh, J.; Alli, L.A.; Shen, B. Phytomedicine in Disease Management: In-Silico Analysis of the Binding Affinity of Artesunate and Azadirachtin for Malaria Treatment. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Analysis. Available online: http://www.strodel.info/index_files/lecture/html/analysis-4.html (accessed on 7 March 2022).
- UCSF Chimera Home Page. Available online: https://www.cgl.ucsf.edu/chimera/ (accessed on 7 March 2022).
- CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Available online: http://sts.bioe.uic.edu/castp/index.html?3trg (accessed on 9 March 2022).
- Tian, H.; Jiang, X.; Tao, P. PASSer: Prediction of allosteric sites server. Mach. Learn. Sci. Technol. 2021, 2, 035015. [Google Scholar] [CrossRef]
- Martínez, L. Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef]
- Friedman, D.J.; Pollak, M.R. APOL1 and Kidney Disease: From Genetics to Biology. Annu. Rev. Physiol. 2020, 82, 323–342. [Google Scholar] [CrossRef]
- Why Structure Prediction Matters|DNASTAR. Available online: https://www.dnastar.com/blog/structural-biology/why-structure-prediction-matters/ (accessed on 8 March 2022).
- Vihinen, M. Functional effects of protein variants. Biochimie 2021, 180, 104–120. [Google Scholar] [CrossRef]
- Udosen, B.; Soremekun, O.; Ekenna, C.; Omotuyi, O.I.; Chikowore, T.; Nashiru, O.; Fatumo, S. In-silico analysis reveals druggable single nucleotide polymorphisms in angiotensin 1 converting enzyme involved in the onset of blood pressure. BMC Res. Notes 2021, 14, 457. [Google Scholar] [CrossRef]
- Subair, T.I.; Akawa, O.B.; Soremekun, O.S.; Olotu, F.A.; Soliman, M.E.S. Insight into the Therapeutic Potential of a Bicyclic Hydroxypyridone Compound 2-[(2,4-Dichlorophenyl)methyl]-7-hydroxy-1,2,3,4-tetrahydro-8H-pyrido[1,2-a]pyrazin-8-one as COMT Inhibitor in the Treatment of Parkinson’s Disease: A Molecular Dynamic Simulation Approach. Chem. Biodivers. 2021, 18, e2100204. [Google Scholar] [CrossRef]
- Haque, S.; Patil, G.; Mishra, A.; Lan, X.; Popik, W.; Malhotra, A.; Skorecki, K.; Singhal, P.C. Effect of APOL1 disease risk variants on APOL1 gene product. Biosci. Rep. 2017, 37, 20160531. [Google Scholar] [CrossRef] [PubMed]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Score | Druggability | Volume | Hydrophobicity | Residues | |
|---|---|---|---|---|---|
| Pocket 1 | 0.023 | 0.002 | 300.261 | 43.667 | N154,L147,L21,K132,L85,C13,V349,S342,Q82,D395,F265,V254,Q134,H130,V338,L258 |
| Pocket 2 | −0.034 | 0.002 | 126.023 | 44.875 | N154,V349,Q239,H360,H241,Q237,Y354,R157,V350,K357,K233,L161,V244,S356,L243,A240,L158,L371,T236,V353,L352 |
| Pocket 3 | −0.037 | 0.001 | 3.333 | 13.167 | V350,A5,Y351,E90,C13,G270,L6,L12,T272,R8,E348,F265,Y354,L266,A269,V9,L347,F344, E92,L86 |
| Energy Component | G1_APIND | G2_APIND |
|---|---|---|
| ∆EvdW (kcal/mol) | −10.23 | −19.084 |
| ∆Eele (kcal/mol) | 20.78 | −40.092 |
| ∆GGB (kcal/mol) | 4.65 | 65.1435 |
| ESURF (kcal/mol) | −3.89 | −5.086 |
| ∆Ggas (kcal/mol) | 6.44 | −99.98 |
| ∆Gsol (kcal/mol) | 4.41 | 60.846 |
| ∆Gbind (kcal/mol) | −10.65 | −20.876 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayanja, R.; Kintu, C.; Diabate, O.; Soremekun, O.; Oluwagbemi, O.O.; Wele, M.; Kalyesubula, R.; Jjingo, D.; Chikowore, T.; Fatumo, S. Molecular Dynamic Simulation Reveals Structure Differences in APOL1 Variants and Implication in Pathogenesis of Chronic Kidney Disease. Genes 2022, 13, 1460. https://doi.org/10.3390/genes13081460
Mayanja R, Kintu C, Diabate O, Soremekun O, Oluwagbemi OO, Wele M, Kalyesubula R, Jjingo D, Chikowore T, Fatumo S. Molecular Dynamic Simulation Reveals Structure Differences in APOL1 Variants and Implication in Pathogenesis of Chronic Kidney Disease. Genes. 2022; 13(8):1460. https://doi.org/10.3390/genes13081460
Chicago/Turabian StyleMayanja, Richard, Christopher Kintu, Oudou Diabate, Opeyemi Soremekun, Olugbenga Oluseun Oluwagbemi, Mamadou Wele, Robert Kalyesubula, Daudi Jjingo, Tinashe Chikowore, and Segun Fatumo. 2022. "Molecular Dynamic Simulation Reveals Structure Differences in APOL1 Variants and Implication in Pathogenesis of Chronic Kidney Disease" Genes 13, no. 8: 1460. https://doi.org/10.3390/genes13081460
APA StyleMayanja, R., Kintu, C., Diabate, O., Soremekun, O., Oluwagbemi, O. O., Wele, M., Kalyesubula, R., Jjingo, D., Chikowore, T., & Fatumo, S. (2022). Molecular Dynamic Simulation Reveals Structure Differences in APOL1 Variants and Implication in Pathogenesis of Chronic Kidney Disease. Genes, 13(8), 1460. https://doi.org/10.3390/genes13081460