
Development of SLAF-Sequence and Multiplex SNaPshot Panels for Population Genetic Diversity Analysis and Construction of DNA Fingerprints for Sugarcane

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction and SLAF Library Construction and Sequencing
2.3. SLAF-Seq Data Grouping and Genotyping
2.4. Data Analysis
2.5. Development of SNaPshot Primers
2.6. SNP Selection and Generation of QR Codes
3. Results
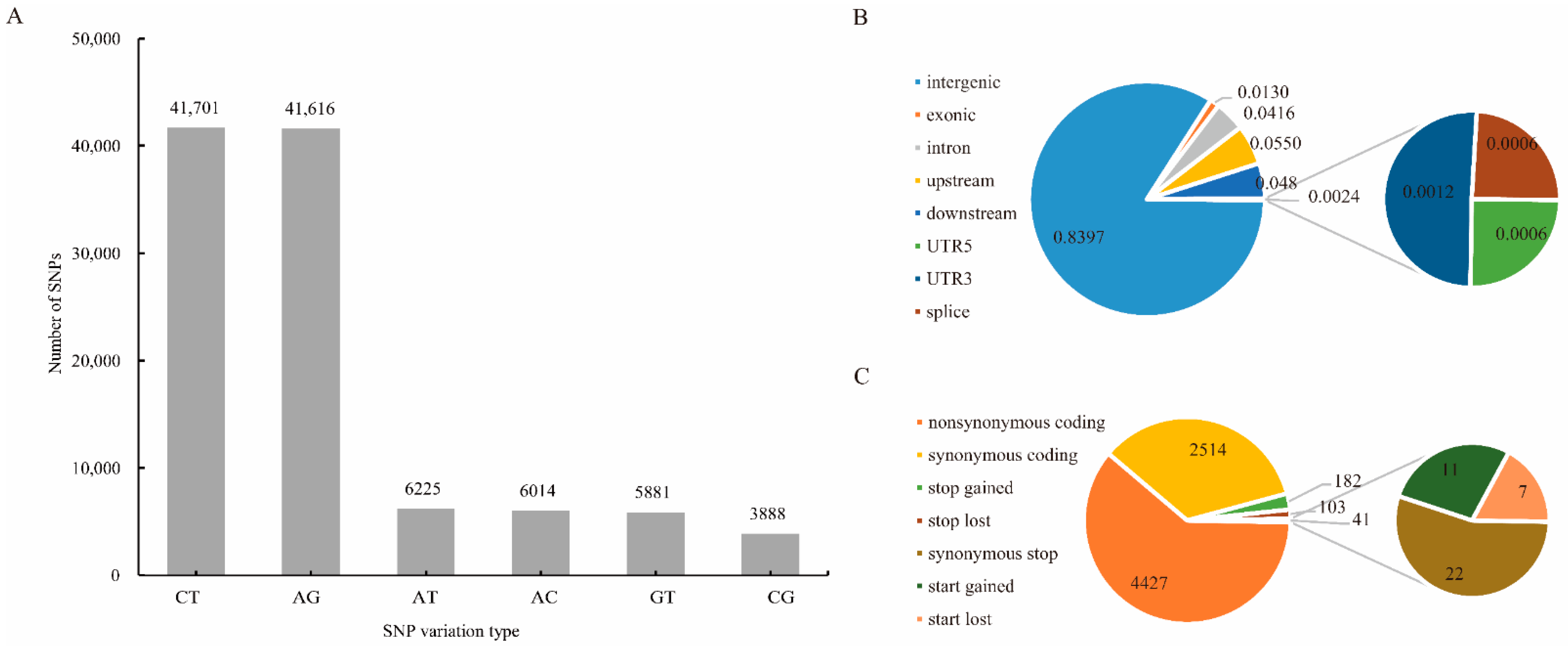
3.1. Sequencing Data Analysis and SNP Identification
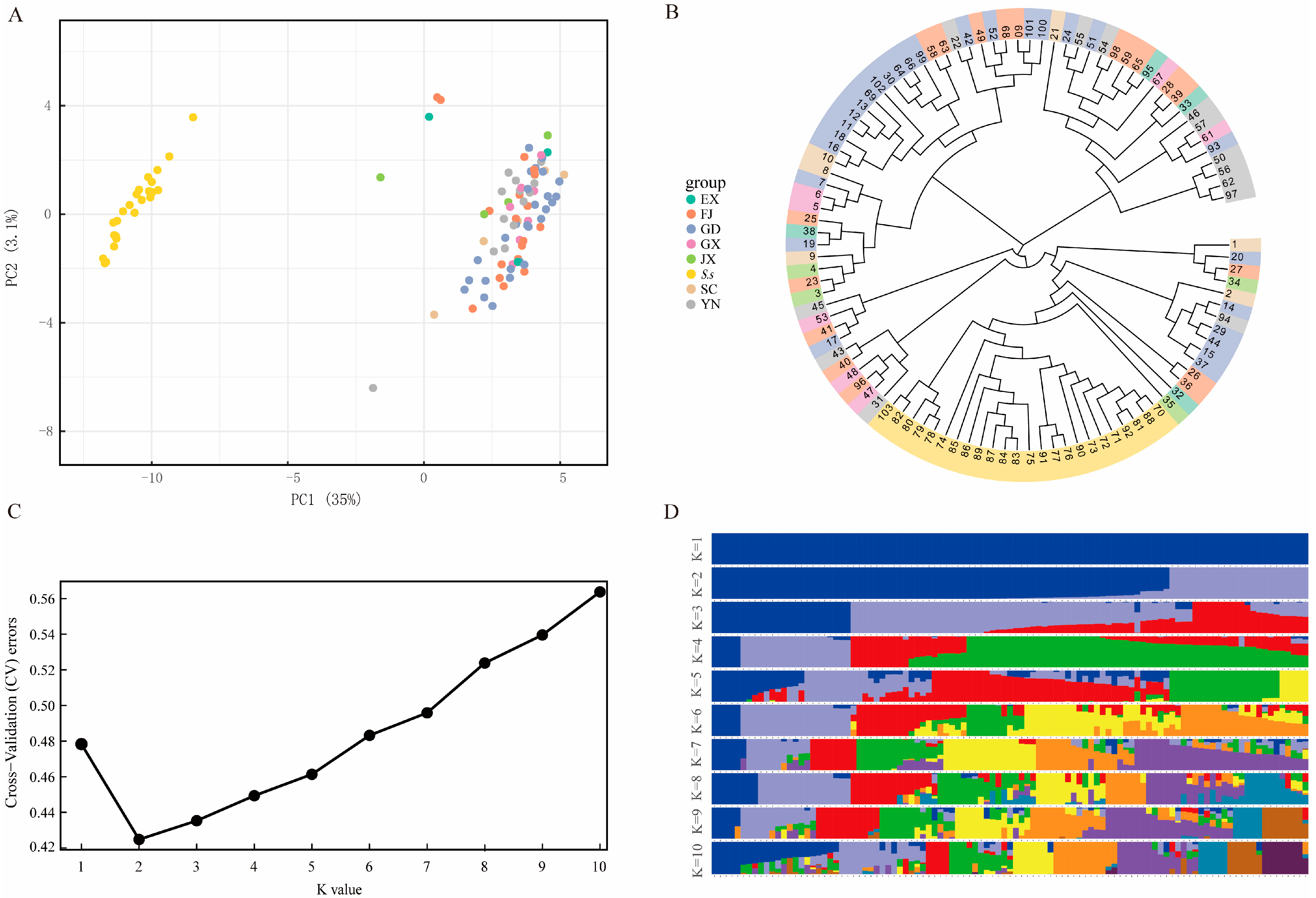
3.2. Genetic Variations and Population Structure
3.3. SNP Primers
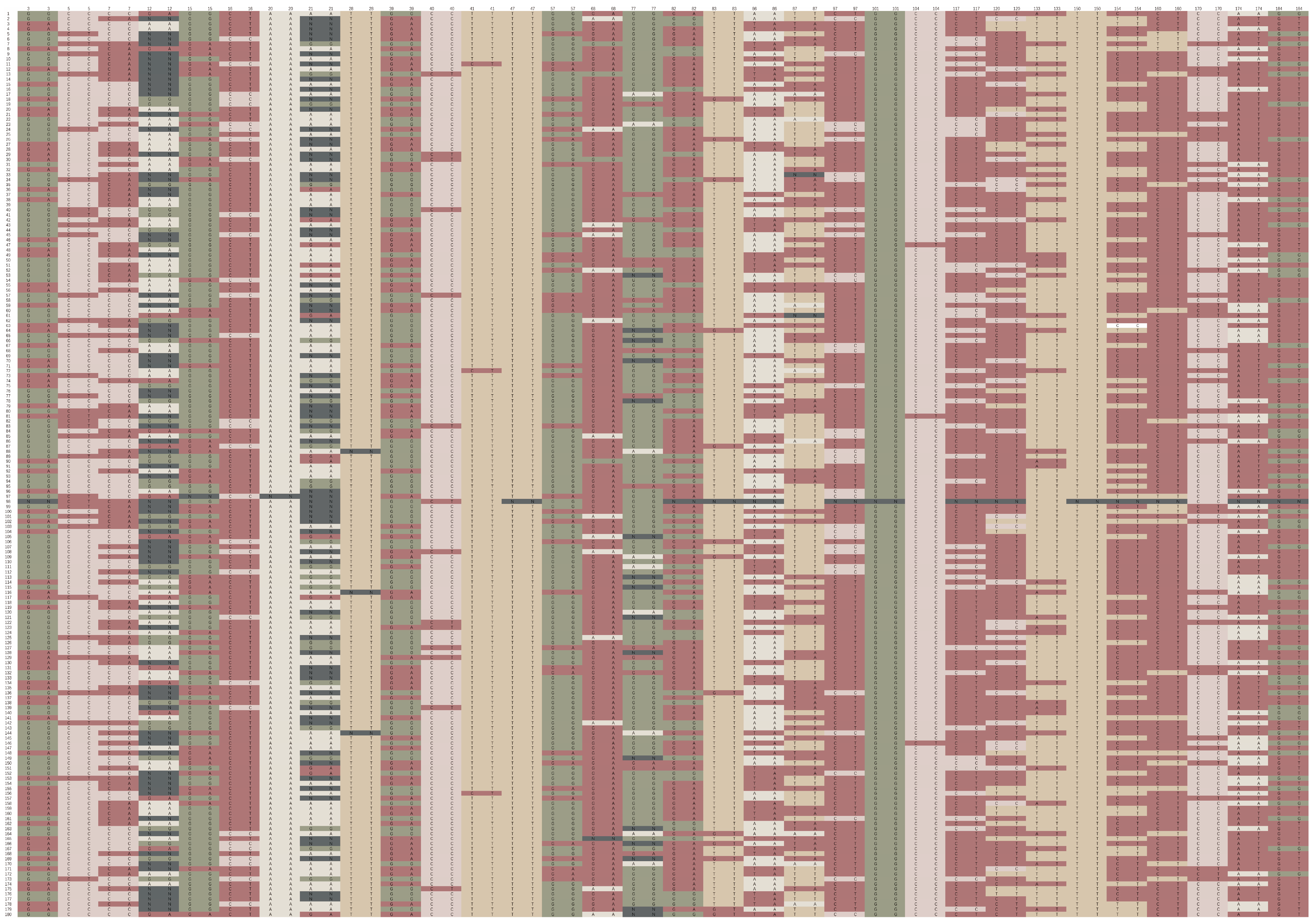
3.4. Construction of DNA Fingerprints
3.5. Ability of Core SNPs to Identify Sugarcane Accessions
4. Discussion
4.1. SNPs Show High Efficiency for Genotyping
4.2. Advantages for SLAF-Sequence and Multiplex SNaPshot Panels Systems
4.3. SNP-Based Genetic Relationships among Sugarcane Accessions
4.4. DNA Fingerprinting Based on SNP Markers
4.5. Application for Fingerprinting and Identification of SNPs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fickett, N.D.; Ebrahimi, L.; Parco, A.P.; Gutierrez, A.V.; Hale, A.L.; Pontif, M.J.; Todd, J.; Kimbeng, C.A.; Hoy, J.W.; Ayala-Silva, T. An enriched sugarcane diversity panel for utilization in genetic improvement of sugarcane. Sci. Rep. 2020, 10, 13390. [Google Scholar] [CrossRef]
- Hoang, N.V.; Furtado, A.; Botha, F.C.; Simmons, B.A.; Henry, R.J. Potential for Genetic Improvement of Sugarcane as a Source of Biomass for Biofuels. Front. Bioeng. Biotechnol. 2015, 3, 182. [Google Scholar] [CrossRef]
- Medeiros, C.; Balsalobre TW, A.; Carneiro, M.S. Molecular diversity and genetic structure of Saccharum complex accessions. PLoS ONE 2020, 15, e0233211. [Google Scholar] [CrossRef]
- Hui, Z.; Pingping, L.; Chaohua, H.; Guoqiang, H.; Liangnian, X.; Zuhu, D.; Muqing, Z.; Xinwang, Z. Research Advances in DNA Molecular Fingerprinting in Sugarcane. SCC 2022, 44, 25–32. [Google Scholar]
- Bohar, R.; Chitkineni, A.; Varshney, R.K. Genetic molecular markers to accelerate genetic gains in crops. Biotechniques 2020, 69, 158–160. [Google Scholar] [CrossRef]
- Dai, S.; Long, Y. Genotyping analysis using an RFLP assay. Methods Mol. Biol. 2015, 1245, 91–99. [Google Scholar]
- Singh, P.; Singh, S.P.; Tiwari, A.K.; Sharma, B.L. Genetic diversity of sugarcane hybrid cultivars by RAPD markers. 3 Biotech 2017, 7, 222. [Google Scholar] [CrossRef]
- Debibakas, S.; Rocher, S.; Garsmeur, O.; Toubi, L.; Roques, D.; D’Hont, A.; Hoarau, J.-Y.; Daugrois, J.H. Prospecting sugarcane resistance to Sugarcane yellow leaf virus by genome-wide association. Theor. Appl. Genet. 2014, 127, 1719–1732. [Google Scholar] [CrossRef]
- Singh, R.B.; Mahenderakar, M.D.; Jugran, A.K.; Singh, R.K.; Srivastava, R.K. Assessing genetic diversity and population structure of sugarcane cultivars, progenitor species and genera using microsatellite (SSR) markers. Gene 2020, 753, 144800. [Google Scholar] [CrossRef]
- Garrido-Cardenas, J.A.; Mesa-Valle, C.; Manzano-Agugliaro, F. Trends in plant research using molecular markers. Planta 2018, 247, 543–557. [Google Scholar] [CrossRef]
- Sun, C.; Dong, Z.; Zhao, L.; Ren, Y.; Zhang, N.; Chen, F. The Wheat 660K SNP array demonstrates great potential for marker-assisted selection in polyploid wheat. Plant Biotechnol. J. 2020, 18, 1354–1360. [Google Scholar] [CrossRef]
- Choudhury, D.R.; Kumar, R.; Singh, K.; Singh, N.K.; Singh, R. Identification of a Diverse Core Set Panel of Rice from the East Coast Region of India Using SNP Markers. Front. Genet. 2021, 12, 726152. [Google Scholar] [CrossRef] [PubMed]
- Arca, M.; Mary-Huard, T.; Gouesnard, B.; Bérard, A.; Bauland, C.; Combes, V.; Madur, D.; Charcosset, A.; Nicolas, S.D. Deciphering the Genetic Diversity of Landraces with High-Throughput SNP Genotyping of DNA Bulks: Methodology and Application to the Maize 50k Array. Front. Plant Sci. 2020, 11, 568699. [Google Scholar] [CrossRef]
- Ganal, M.W.; Altmann, T.; Röder, M.S. SNP identification in crop plants. Curr. Opin. Plant Biol. 2009, 12, 211–217. [Google Scholar] [CrossRef]
- Yang, H.; Li, C.; Lam, H.-M.; Clements, J.; Yan, G.; Zhao, S. Sequencing consolidates molecular markers with plant breeding practice. Theor. Appl. Genet. 2015, 128, 779–795. [Google Scholar] [CrossRef]
- You, Q.; Yang, X.; Peng, Z.; Islam, S.; Sood, S.; Luo, Z.; Comstock, J.; Xu, L.; Wang, J. Development of an Axiom Sugarcane100K SNP array for genetic map construction and QTL identification. Theor. Appl. Genet. 2019, 132, 2829–2845. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Zhu, Z.; Sun, B.; Lei, J. Specific-Locus Amplified Fragment Sequencing (SLAF-Seq) as High-Throughput SNP Genotyping Methods. Methods Mol. Biol. 2021, 2264, 75–87. [Google Scholar]
- Amanullah, S.; Osae, B.A.; Yang, T.; Abbas, F.; Liu, S.; Liu, H.; Wang, X.; Gao, P.; Luan, F. Mapping of genetic loci controlling fruit linked morphological traits of melon using developed CAPS markers. Mol. Biol. Rep. 2022, 49, 5459–5472. [Google Scholar] [CrossRef] [PubMed]
- Bollinedi, H.; Singh, N.; Krishnan, S.G.; Vinod, K.K.; Bhowmick, P.K.; Nagarajan, M.; Ellur, R.K.; Singh, A.K. A novel LOX3-null allele (lox3-b) originated in the aromatic Basmati rice cultivars imparts storage stability to rice bran. Food Chem. 2022, 369, 130887. [Google Scholar] [CrossRef]
- Hong, S.; Choi, S.R.; Kim, J.; Jeong, Y.M.; Kim, J.S.; Ahn, C.H.; Kwon, S.Y.; Lim, Y.P.; Shin, A.Y.; Kim, Y.M. Identification of accession-specific variants and development of KASP markers for assessing the genetic makeup of Brassica rapa seeds. BMC Genom. 2022, 23, 326. [Google Scholar] [CrossRef]
- Manimekalai, R.; Suresh, G.; Kurup, H.G.; Athiappan, S.; Kandalam, M. Role of NGS and SNP genotyping methods in sugarcane improvement programs. Crit. Rev. Biotechnol. 2020, 40, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, S.; Huang, Y.; Zhang, B.; Xu, Y.; Zhang, G.; Lakshmanan, P.; Yang, R.; Zhou, H.; Huang, D.; et al. Quantitative Trait Loci Mapping and Development of KASP Marker Smut Screening Assay Using High-Density Genetic Map and Bulked Segregant RNA Sequencing in Sugarcane (Saccharum spp.). Front. Plant Sci. 2021, 12, 796189. [Google Scholar] [CrossRef] [PubMed]
- Mehta, B.; Daniel, R.; Phillips, C.; McNevin, D. Forensically relevant SNaPshot(®) assays for human DNA SNP analysis: A review. Int. J. Leg. Med. 2017, 131, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Jung, J.Y.; Choi, S.; Seol, I.; Moon, S.; Hwang, I.K. Single Nucleotide Polymorphism Assay for Genetic Identification of Lophophora williamsii. J. Forensic Sci. 2020, 65, 2117–2120. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kim, H.; Bae, P.K.; Lee, S.; Yang, S.; Kim, J. A single snapshot multiplex immunoassay platform utilizing dense test lines based on engineered beads. Biosens. Bioelectron. 2021, 190, 113388. [Google Scholar] [CrossRef]
- Törjék, O.; Berger, D.; Meyer, R.C.; Müssig, C.; Schmid, K.J.; Sörensen, T.R.; Weisshaar, B.; Mitchell-Olds, T.; Altmann, T. Establishment of a high-efficiency SNP-based framework marker set for Arabidopsis. Plant J. 2003, 36, 122–140. [Google Scholar] [CrossRef]
- Deleu, W.; Esteras, C.; Roig, C.; González-To, M.; Fernández-Silva, I.; Gonzalez-Ibeas, D.; Blanca, J.; Aranda, M.A.; Arús, P.; Nuez, F.; et al. A set of EST-SNPs for map saturation and cultivar identification in melon. BMC Plant Biol. 2009, 9, 90. [Google Scholar] [CrossRef]
- Zhang, C.; Tan, L.; Wang, L.; Wei, K.; Cheng, H. Study of SNaPshot Detect SNP Markers in Tea Plant. J. Tea Sci. 2014, 34, 180–187. [Google Scholar]
- Budeguer, F.; Enrique, R.; Perera, M.F.; Racedo, J.; Castagnaro, A.P.; Noguera, A.S.; Welin, B. Genetic Transformation of Sugarcane, Current Status and Future Prospects. Front. Plant Sci. 2021, 12, 768609. [Google Scholar] [CrossRef]
- Button, P. The international union for the protection of new varieties of plants (upov) recommendations on variety denominations. Acta Hortic. 2008, 799, 191–200. [Google Scholar] [CrossRef]
- Jones, H.; Norris, C.; Smith, D.; Cockram, J.; Lee, D.; O’Sullivan, D.M.; Mackay, I. Evaluation of the use of high-density SNP genotyping to implement UPOV Model 2 for DUS testing in barley. Theor. Appl. Genet. 2013, 126, 901–911. [Google Scholar] [CrossRef]
- Jones, H.; Mackay, I. Implications of using genomic prediction within a high-density SNP dataset to predict DUS traits in barley. Theor. Appl. Genet. 2015, 128, 2461–2470. [Google Scholar] [CrossRef]
- Tian, H.L.; Wang, F.G.; Zhao, J.R.; Yi, H.M.; Wang, L.; Wang, R.; Yang, Y.; Song, W. Development of maizeSNP3072, a high-throughput compatible SNP array, for DNA fingerprinting identification of Chinese maize varieties. Mol. Breed. 2015, 35, 136. [Google Scholar] [CrossRef]
- Raatz, B.; Mukankusi, C.; Lobaton, J.D.; Male, A.; Chisale, V.; Amsalu, B.; Fourie, D.; Mukamuhirwa, F.; Muimui, K.; Mutari, B.; et al. Analyses of African common bean (Phaseolus vulgaris L.) germplasm using a SNP fingerprinting platform: Diversity, quality control and molecular breeding. Genet. Resour. Crop. Evol. 2019, 66, 707–722. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, H.; Xiang, X.; Yang, A.; Feng, Q.; Dai, P.; Li, Y.; Jiang, X.; Liu, G.; Zhang, X. Construction of a SNP Fingerprinting Database and Population Genetic Analysis of Cigar Tobacco Germplasm Resources in China. Front. Plant Sci. 2021, 12, 618133. [Google Scholar] [CrossRef]
- Abdel-Latif, A.; Osman, G. Comparison of three genomic DNA extraction methods to obtain high DNA quality from maize. Plant Methods 2017, 13, 1. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef]
- Davey, J.W.; Cezard, T.; Fuentes-Utrilla, P.; Eland, C.; Gharbi, K.; Blaxter, M.L. Special features of RAD Sequencing data: Implications for genotyping. Mol. Ecol. 2013, 22, 3151–3164. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Env. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Chatterji, S.; Pachter, L. Reference based annotation with GeneMapper. Genome Biol. 2006, 7, R29. [Google Scholar] [CrossRef]
- Fang, H.; Liu, H.; Ma, R.; Liu, Y.; Li, J.; Yu, X.; Zhang, H.; Yang, Y.; Zhang, G. Genome-wide assessment of population structure and genetic diversity of Chinese Lou onion using specific length amplified fragment (SLAF) sequencing. PLoS ONE 2020, 15, e0231753. [Google Scholar] [CrossRef]
- Su, W.; Wang, L.; Lei, J.; Chai, S.; Liu, Y.; Yang, Y.; Yang, X.; Jiao, C. Genome-wide assessment of population structure and genetic diversity and development of a core germplasm set for sweet potato based on specific length amplified fragment (SLAF) sequencing. PLoS ONE 2017, 12, e0172066. [Google Scholar] [CrossRef]
- Li, C.; Liu, M.; Sun, F.; Zhao, X.; He, M.; Li, T.; Lu, P.; Xu, Y. Genetic Divergence and Population Structure in Weedy and Cultivated Broomcorn Millets (Panicum miliaceum L.) Revealed by Specific-Locus Amplified Fragment Sequencing (SLAF-Seq). Front. Plant Sci. 2021, 12, 688444. [Google Scholar] [CrossRef]
- Jagtap, A.B.; Vikal, Y.; Johal, G.S. Genome-Wide Development and Validation of Cost-Effective KASP Marker Assays for Genetic Dissection of Heat Stress Tolerance in Maize. Int. J. Mol. Sci. 2020, 21, 7386. [Google Scholar] [CrossRef]
- Grewal, S.; Guwela, V.; Newell, C.; Yang, C.-Y.; Ashling, S.; Scholefield, D.; Hubbart-Edwards, S.; Burridge, A.; Stride, A.; King, I.P.; et al. Generation of Doubled Haploid Wheat-Triticum urartu Introgression Lines and Their Characterisation Using Chromosome-Specific KASP Markers. Front. Plant Sci. 2021, 12, 643636. [Google Scholar] [CrossRef]
- Yang, S.; Yu, W.; Wei, X.; Wang, Z.; Zhao, Y.; Zhao, X.; Tian, B.; Yuan, Y.; Zhang, X.; Juan, Y. An extended KASP-SNP resource for molecular breeding in Chinese cabbage(Brassica rapa L. ssp. pekinensis). PLoS ONE 2020, 15, e0240042. [Google Scholar] [CrossRef]
- He, C.; Holme, J.; Anthony, J. SNP genotyping: The KASP assay. Methods Mol. Biol. 2014, 1145, 75–86. [Google Scholar]
- Dai, Z. SNP Group Study of Gastric Cancer Susceptibility in Jiangsu Han Population Based on SNaPshot Technology. Master’s Thesis, Jiangsu University, Zhenjiang, China, 2021. [Google Scholar]
- Mahadevaiah, C.; Appunu, C.; Aitken, K.; Suresha, G.S.; Vignesh, P.; Swamy, H.K.M.; Valarmathi, R.; Hemaprabha, G.; Alagarasan, G.; Ram, B. Genomic Selection in Sugarcane: Current Status and Future Prospects. Front. Plant Sci. 2021, 12, 708233. [Google Scholar] [CrossRef]
- Pan, Y.B.; Tew, T.L.; Schnell, R.J.; Viator, R.P.; Richard, E.P.; Grisham, M.P.; White, W.H. Microsatellite DNA marker-assisted selection of Saccharum spontaneum cytoplasm-derived germplasm. Sugar Tech. 2006, 8, 23–29. [Google Scholar] [CrossRef]
- Pan, Y.B. Databasing Molecular Identities of Sugarcane (Saccharum spp.) Clones Constructed with Microsatellite (SSR) DNA Markers. Am. J. Plant Sci. 2010, 1, 87–94. [Google Scholar] [CrossRef]
- Ali, A. Construction of DNA Fingerprinting and Analysis of Genetic Diversity and Population Structure with SSR Markers for Major Released Varieties and Wild Germplasm Resources of Sugarcane from China. Ph.D. Thesis, Fujian Agriculture and Forestry University, Fuzhou, China, 2018. [Google Scholar]
- Khlestkina, E.K.; Salina, E.A. SNP markers: Methods of analysis, ways of development, and comparison on an example of common wheat. Genetika 2006, 42, 725–736. [Google Scholar] [CrossRef]
- Corrado, G.; Piffanelli, P.; Caramante, M.; Coppola, M.; Rao, R. SNP genotyping reveals genetic diversity between cultivated landraces and contemporary varieties of tomato. BMC Genom. 2013, 14, 835. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Lin, P.; Liu, Y.; Huang, C.; Huang, G.; Jiang, H.; Xu, L.; Zhang, M.; Deng, Z.; Zhao, X. Development of SLAF-Sequence and Multiplex SNaPshot Panels for Population Genetic Diversity Analysis and Construction of DNA Fingerprints for Sugarcane. Genes 2022, 13, 1477. https://doi.org/10.3390/genes13081477
Zhang H, Lin P, Liu Y, Huang C, Huang G, Jiang H, Xu L, Zhang M, Deng Z, Zhao X. Development of SLAF-Sequence and Multiplex SNaPshot Panels for Population Genetic Diversity Analysis and Construction of DNA Fingerprints for Sugarcane. Genes. 2022; 13(8):1477. https://doi.org/10.3390/genes13081477
Chicago/Turabian StyleZhang, Hui, Pingping Lin, Yanming Liu, Chaohua Huang, Guoqiang Huang, Hongtao Jiang, Liangnian Xu, Muqing Zhang, Zuhu Deng, and Xinwang Zhao. 2022. "Development of SLAF-Sequence and Multiplex SNaPshot Panels for Population Genetic Diversity Analysis and Construction of DNA Fingerprints for Sugarcane" Genes 13, no. 8: 1477. https://doi.org/10.3390/genes13081477
APA StyleZhang, H., Lin, P., Liu, Y., Huang, C., Huang, G., Jiang, H., Xu, L., Zhang, M., Deng, Z., & Zhao, X. (2022). Development of SLAF-Sequence and Multiplex SNaPshot Panels for Population Genetic Diversity Analysis and Construction of DNA Fingerprints for Sugarcane. Genes, 13(8), 1477. https://doi.org/10.3390/genes13081477