Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that can rarely affect young individuals. Juvenile ALS (JALS) is defined for individuals with an onset of the disease before the age of 25. The contribution of genetics to ALS pathology is a field of growing interest. One of the differences between adult-onset ALS and JALS is their genetic background, with a higher contribution of genetic causes in JALS. We report a patient with JALS and a pathogenic variant in the TARDBP gene (c.1035C > G; p.Asn345Lys), previously reported only in adult-onset ALS, and with an atypical phenotype of marked upper motor neuron predominance. In addition, the proband presented an additional variant in the NEK1 gene, c.2961C > G (p.Phe987Leu), which is classified as a variant of unknown significance. Segregation studies showed a paternal origin of the TARDBP variant, while the variant in NEK1 was inherited from the mother. We hypothesize that the NEK1 variant acts as a disease modifier and suggests the possibility of a functional interaction between both genes in our case. This hypothesis could explain the peculiarities of the phenotype, penetrance, and the age of onset. This report highlights the heterogeneity of the phenotypic presentation of ALS associated with diverse pathogenic genetic variants.
1. Introduction
Amyotrophic lateral sclerosis (ALS), classically regarded as the most common motor neuron disease, is now recognized as a multisystemic neurodegenerative disorder with great heterogeneity in its clinical and pathophysiological aspects [1]. Research in the last decades has revealed multiple pathological pathways involved, including various genetic aspects and the description of several causative genes [2,3]. Although it predominantly affects adults, in a small percentage of cases, young individuals are affected. Juvenile ALS (JALS) is typically defined for individuals with an onset of the disease before the age of 25 [4,5].
One of the main differences between adult-onset ALS and JALS is their genetic background, with a higher contribution of genetic causes detected in JALS (40%) than in adult-onset ALS (around 10%) [2,6,7]. The frequencies of associated genes are also different between the two forms of onset. A recent review of JALS [7] identified FUS (fused in sarcoma), SETX (Senataxin), and ALS2 (Alsin) as the most common genes reported in the literature. In contrast, pathogenic variants in genes commonly associated with adult forms, such as SOD1 (copper–zinc superoxide dismutase) or TARDBP (transactive response DNA-binding protein), were reported less frequently, and C9orf72 (chromosome 9 open reading frame 72), the most prevalent inherited gene in adult disease, has not been reported in childhood [7,8].
We report herein a patient with JALS and a pathogenic variant in the TARDBP gene (c.1035C > G, p.Asn345Lys), previously reported only in adult-onset ALS, and with an atypical phenotype of marked upper motor neuron predominance. In addition, we performed a literature review of JALS cases associated with variants in the TARDBP gene to discuss the phenotype.
2. Patient and Method
2.1. Clinical Presentation
A 24-year-old male patient of Spanish origin with no relevant medical history presented to our neuromuscular disease unit with a progressive disorder of rigidity and weakness of the lower limbs. The symptoms had started 3 months earlier, initially affecting the left lower limb and causing difficulty in walking. The first neurological examination revealed signs of upper motor neuron involvement with hyperreflexia and spasticity in all four limbs, a sustained left ankle clonus, bilateral Babinski sign, and bilateral Hoffman sign. There was a slight distal motor deficit in the left lower limb (4/5 on tibialis anterior according to the Medical Research Council scale) and a spastic gait pattern with greater involvement of the left side. No evidence of lower motor neuron impairment, bulbar involvement, or sensory disturbance was observed.
Brain and spinal magnetic resonance imaging (MRI) showed T2-weighted and fluid-attenuated inversion recovery (FLAIR) increased signal intensities at the pyramidal tracts bilaterally, suggestive of corticospinal pathway degeneration (Figure 1). The electrophysiological tests showed evidence of lower motor neuron involvement, with active denervation in lower extremity muscles in needle electromyography (tibialis anterior, vastus medialis, and gastrocnemius bilaterally) and a conduction defect in the corticospinal pathway in both upper and lower extremities using transcranial magnetic stimulation. Cerebrospinal fluid analysis was entirely normal. Further comprehensive laboratory workup ruled out other inflammatory or infectious causes.
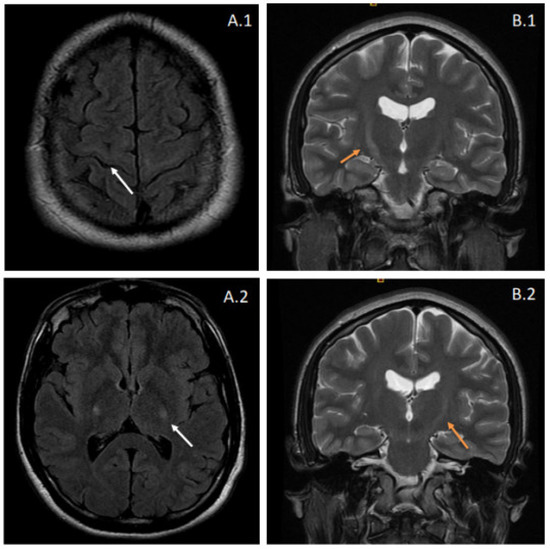
Figure 1.
Brain MRI from the proband. Images (A.1,A.2) correspond to fluid-attenuated inversion recovery (FLAIR) sequences, showing hyperintensity affecting the pyramidal tracts bilaterally (white arrows). These features suggest degeneration of the corticospinal pathway and can also be observed in coronal T2-weighted images (B.1,B.2) as increased signal intensities at the internal capsule. The latter radiologic appearance is also known as the “Wine Glass” sign.
The evolution in the following months showed a fast progression of the disorder. Slight signs of lower motor neuron lesion appeared in the spinal territory with sporadic fasciculations in the upper and lower extremities. The pyramidal syndrome spread to the upper extremities and bulbar region, and the patient developed spastic dysarthria and dysphagia. The distal weakness in the left lower extremity and spasticity progressively worsened, losing independent walking ability eight months after onset. A diagnosis of clinically probable ALS was made according to the revised El Escorial criteria [9] and treatment with riluzole 50 mg twice was initiated.
There was no family history of motor neuron diseases, dementia, or psychiatric disorders. The patient was an only child born of non-consanguineous parents. His father was 61 years old, and his mother was 62 years old at the moment of the evaluation. Both parents were examined as well and were clinically unaffected, and had no health complaints of neurologic nature. Considering the clinical diagnosis and age of onset, genetic studies were performed.
2.2. Genetic Results
SOD1 and C9orf72 were studied first, given that they are the most prevalent genes associated with ALS in the European population [2] and the emerging therapies for ALS due to pathogenic variants in these genes (NCT04972487, NCT049937557). Sanger sequencing of SOD1 showed no pathogenic variants. The pathogenic repeat expansion of C9orf72 was discarded by polymerase chain reaction (PCR) amplification, subsequent analysis of the fragments by capillary electrophoresis, and confirmation of the GGGGCC hexanucleotide expansion by repeat-primed PCR (RP-PCR).
Subsequently, we performed exome sequencing prioritizing variants in genes associated with ALS and frontotemporal dementia (FTD) as well as genes based on appropriate HPO terms. These terms and the analyzed genes are listed as Supplementary Materials. A heterozygous variant c.1035C > G (p.Asn345Lys) was detected in the TARDBP gene (NM_007375.3). This variant was considered according to the American College of Medical Genetics (PS1, PS4, PM1, PM2) [10]. It has not been previously described in general population databases (GnomAD), but has been in several patients with this condition [11,12,13]. An additional heterozygous variant c.2961C > G (p.Phe987Leu) in the NEK1 gene (NM_012224.2) was identified and classified as a variant of uncertain significance.
Segregation analysis in the parents showed that the pathogenic TARDBP variant was inherited from the father, whereas the NEK1 variant was inherited from the mother.
3. Discussion
In this case report, we present a patient with genetically determined ALS caused by a pathogenic variant in the TARDBP gene associated with a remarkably atypical phenotype mainly for two reasons: early age of onset, compatible with JALS, and the significant predominance of upper motor neuron involvement. The patient carried an additional variant in NEK1, classified as of uncertain significance.
The TARDBP gene encodes for the TAR DNA-binding protein-43 (TDP-43), a highly conserved DNA/RNA binding protein with a ubiquitous expression that belongs to the heterogeneous nuclear ribonucleoprotein (hnRNP) family. In 2006, intraneuronal ubiquitinated cytoplasmic inclusions, observed in numerous pathological ALS and FTD specimens, were found to have TDP-43 as a major component [14,15]. These neuronal aggregates of hyperphosphorylated TDP-43 in the brain and spinal cord are considered a histopathological hallmark of the disease and are identified in the vast majority of ALS patients, including sporadic forms and those carrying pathogenic variants (with a few exceptions, such as SOD1 or FUS genes). TDP-43 exerts numerous cellular functions, and multiple mechanisms have been implicated in its pathogenesis [16]. It plays a central role in RNA metabolism, regulating transcription, translation, splicing, mRNA stability and transport, and microRNA maturation. It is also relevant in neuronal defense mechanisms against oxidative stress, and the proper functioning of mitochondrial respiratory chain pathways. Reflecting these functions, mitochondrial localization motifs (M1-M3) and RNA recognition motifs (RRM1 and RRM2) are found in their 414 amino acid structure (Figure 2). In addition, there are sequences of the nuclear localization signal (NLS) and nuclear export signal (NES) because, in physiological conditions, TDP-43 translocates between the nucleus and the cytoplasm to perform these functions [17]. In 2008, mutations in TARDBP were first described as a dominant cause of genetic ALS [18,19,20], and to date, more than 48 pathogenic variants have been identified [2]. The majority of these variants are missense and located in the C-terminal region. A low-complexity domain with glycine-rich and glutamine/asparagine-rich (Q/N) sequences is situated here and is considered a prion-like protein (PLP) domain [16].
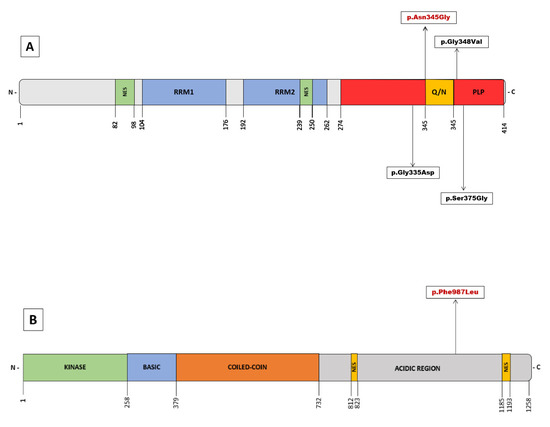
Figure 2.
(A) Overview of pathogenic variants in TARDBP identified in Juvenil ALS. Schematic domain structure of TAR DNA-binding protein 43 (TDP-43). (B) Schematic representation of the NEK1 protein structure with the identified variant of uncertain significance. The numbers represent amino acid lengths.
The variant identified in our patient had only been described in patients with an adult-onset disease [11,12,13], comprising a total of 4 patients from 3 different families, all of them of Asian origin and with symptom onset after 60 years. A variant at the same position but with a different amino acid substitution (c.1035C > A) is a well-known pathogenic variant in adult-onset ALS, included in most genetic databases and used in various molecular models of the disorder [21,22]. TARDBP-associated ALS cases develop a classic ALS phenotype with a mean age of onset of 53 years but with wide variability in both onset and duration [23,24]. The most distinctive phenotypic feature is a more frequent onset in the upper extremities, but with a similar evolution to the sporadic ALS, including the extension to other regions and bulbar involvement. To our knowledge, only four cases of JALS associated with three different variants in the TARDBP gene have been described [25,26,27,28] (Table 1). An overview of the clinical data of these cases reveals the following. In three cases, the clinical onset was in the upper limbs. In all cases, the initial motor symptoms and those causing the most disability corresponded to predominantly lower motor neuron symptoms, such as weakness and amyotrophy. Disease duration ranged from 24 to 48 months, although one patient remained alive at the publication date (more than 120 months from onset). In common with most adult-onset pathogenic variants, they are dominant missense variants and are located in the C-terminal region (Figure 2A). Two of these pathogenic variants associated with TARDBP-JALS have also been reported in adult-onset individuals [29,30,31,32,33,34,35]. Despite the limitation of the small number of cases and the absence of detailed clinical descriptions, a comparison of the phenotypes does not reveal a major preference for upper motor neuron involvement as seen in our patient.

Table 1.
Clinical characteristics of Juvenile ALS TARDBP-associated patients. FALS: familial ALS. UL: upper limbs. LL: lower limbs. LMN: lower motor neuron. UMN: upper motor neuron. CI: cognitive impairment.
In the present case, the variant was inherited from the father’s proband, which shows no clinical or neurophysiological evidence of motor neuron disease or cognitive impairment. Although other members of the paternal family could not be studied, a complete family pedigree history was obtained with no significant findings. It raises the possibility of incomplete or age-dependent penetrance, as has been previously described in the literature. The main series of patients with TARDBP pathogenic variants show a median age at disease onset of 53.5 years +/− 12.3 SD [23]. Although there is no detailed information on the overall penetrance of the pathogenic variants in this gene, there are reports of incomplete penetrance in FTD-TARDBP [36] and ALS. The most comprehensive research on the risk of ALS in patients carrying a pathogenic variant in TARDBP has been carried out in the Sardinian population, given the high prevalence of the p.(Ala382Thr) pathogenic variant. Several studies in this population revealed a penetrance approximately of 60% at 70 years of age but with differences between sexes, pointing to a higher penetrance in men (74–80%) than in women (42.5–66%) [37].
In addition to the pathogenic variant in TARDBP, the proband presented an additional variant in the NEK1 gene, p.(Phe987Leu), which is classified as a variant of unknown significance, and its role in the pathogenesis of our patient is uncertain. NEK1 was recently described as a gene associated with ALS [38,39] with both loss of function and missense variants postulated as risk factors. NEK1 encodes for a serine/threonine protein-kinase with functions in cell cycle control, DNA damage repair, cilia regulation, and apoptosis. Loss of function variants in NEK1 have been associated with an elevated risk of developing ALS [40,41,42]. This variant is located in the acidic region proximal to the C-terminal region but not in any known specific domain (Figure 2B). The distribution of the known pathogenic or likely pathogenic variants in the NEK1 gene does not show clustering in hot spots but appears to be distributed throughout its structure [43]. This role as a risk factor suggests a synergistic effect with other players, including other ALS-associated genes, thus supporting the hypothesis of oligogenic inheritance in ALS. Some studies have identified a high frequency of NEK1 variants and other ALS-associated genes in the same patient [43]. Although the pathogenic role of missense variants remains more controversial, recent studies provide evidence for a possible association of ALS with missense variants in NEK1 gene [43,44]. For example, a patient carrying a pathogenic variant in the TARDBP gene also presenting the missense variant p.(Arg261His) in NEK1, considered as a phenotype modifier with earlier disease onset, has been described [41]. To our knowledge, beyond the enrichment of pathogenic variants in ALS-associated genes in carriers of NEK1 variants, other evidence of a functional interaction between both genes has not been described in the literature. In our case, segregation studies showed that the pathogenic TARDBP variant was inherited from the father, whereas the rare variant in NEK1 was inherited from the mother. This finding is consistent with a previous hypothesis suggesting a second-hit model in which NEK1 variants act as disease modifiers and suggests the possibility of an epistatic effect of both genes in this particular case. This hypothesis could explain the peculiarities of the phenotype, penetrance, intrafamilial variability, and especially the age of onset seen in our patient. However, this statement should be considered a preliminary hypothesis that requires further study.
4. Conclusions
This report highlights the heterogeneity of the phenotypic presentation of ALS associated with diverse pathogenic genetic variants. Furthermore, this case provides further evidence that pathogenic variants in the TARDBP gene may be an infrequent cause of JALS.
The view of ALS as a disease with multistep pathophysiology emphasizes the accumulation of various hits in different pathological pathways. In this sense, a polygenic inheritance could explain part of the disease variability, and the expansion of new genetic techniques in the coming years will offer new insights into this field.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes13081483/s1, Human Phenotype Ontology (HPO) terms used and list of genes analysed in blood DNA next-generation exome sequencing study.
Author Contributions
Conceptualization and design of the work, D.S.-T., J.L.R.-V., A.L., M.S., J.S. and R.J.-M.; data acquisition, D.S.-T., J.L.R.-V., E.R.-M., M.C.-S., A.L., M.S. and J.S.; writing and original draft preparation, D.S.-T. and J.L.R.-V.; review and editing, D.S.-T., J.L.R.-V., E.R.-M., M.C.-S., A.L., M.S., J.S., N.R., E.G.-A. and R.J.-M.; creation and edition of images and figures, D.S.-T. and J.L.R.-V.; genetic methodology, E.R.-M. and M.C.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent has been obtained from the patient(s) to publish this paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Masson, G.L. Genetics of Amyotrophic Lateral Sclerosis: A Review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Orban, P.; Devon, R.S.; Hayden, M.R.; Leavitt, B.R. Chapter 15 Juvenile amyotrophic lateral sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2007; Volume 82, pp. 301–312. ISBN 978-0-444-51894-1. [Google Scholar]
- Sreedharan, J.; Brown, R.H. Juvenile Amyotrophic Lateral Sclerosis. In Neuromuscular Disorders of Infancy, Childhood, and Adolescence; Elsevier: Amsterdam, The Netherlands, 2015; pp. 146–159. ISBN 978-0-12-417044-5. [Google Scholar]
- Kacem, I.; Sghaier, I.; Bougatef, S.; Nasri, A.; Gargouri, A.; Ajroud-Driss, S.; Gouider, R. Epidemiological and Clinical Features of Amyotrophic Lateral Sclerosis in a Tunisian Cohort. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Lehky, T.; Grunseich, C. Juvenile Amyotrophic Lateral Sclerosis: A Review. Genes 2021, 12, 1935. [Google Scholar] [CrossRef] [PubMed]
- Esselin, F.; Mouzat, K.; Polge, A.; Juntas-Morales, R.; Pageot, N.; De la Cruz, E.; Bernard, E.; Lagrange, E.; Danel, V.; Alphandery, S.; et al. Clinical Phenotype and Inheritance in Patients with C9ORF72 Hexanucleotide Repeat Expansion: Results from a Large French Cohort. Front. Neurosci. 2020, 14, 316. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hara, K.; Ishihara, T.; Onodera, O.; Ishiguro, H. A New Japanese Amyotrophic Lateral Sclerosis Family with TARDBP (TDP-43) Mutation. Neurol. Clin. Neurosci. 2019, 7, 101–102. [Google Scholar] [CrossRef]
- Takeda, T.; Iijima, M.; Shimizu, Y.; Yoshizawa, H.; Miyashiro, M.; Onizuka, H.; Yamamoto, T.; Nishiyama, A.; Suzuki, N.; Aoki, M.; et al. P.N345K Mutation in TARDBP in a Patient with Familial Amyotrophic Lateral Sclerosis: An Autopsy Case. Neuropathology 2019, 39, 286–293. [Google Scholar] [CrossRef]
- Leventoux, N.; Morimoto, S.; Hara, K.; Nakamura, S.; Ozawa, F.; Mitsuzawa, S.; Akiyama, T.; Nishiyama, A.; Suzuki, N.; Warita, H.; et al. Generation of an ALS Human IPSC Line KEIOi001-A from Peripheral Blood of a Charcot Disease-Affected Patient Carrying TARDBP p.N345K Heterozygous SNP Mutation. Stem Cell Res. 2020, 47, 101896. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 Proteinopathies: A New Wave of Neurodegenerative Diseases. J. Neurol. Neurosurg. Psychiatry 2021, 92, 86–95. [Google Scholar] [CrossRef] [PubMed]
- François-Moutal, L.; Perez-Miller, S.; Scott, D.D.; Miranda, V.G.; Mollasalehi, N.; Khanna, M. Structural Insights Into TDP-43 and Effects of Post-Translational Modifications. Front. Mol. Neurosci. 2019, 12, 301. [Google Scholar] [CrossRef]
- Gitcho, M.A.; Baloh, R.H.; Chakraverty, S.; Mayo, K.; Norton, J.B.; Levitch, D.; Hatanpaa, K.J.; White, C.L.; Bigio, E.H.; Caselli, R.; et al. TDP-43 A315T Mutation in Familial Motor Neuron Disease. Ann. Neurol. 2008, 63, 535–538. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP Mutations in Individuals with Sporadic and Familial Amyotrophic Lateral Sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 Is Intrinsically Aggregation-Prone, and Amyotrophic Lateral Sclerosis-Linked Mutations Accelerate Aggregation and Increase Toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Budini, M.; Romano, V.; Avendaño-Vázquez, S.E.; Bembich, S.; Buratti, E.; Baralle, F.E. Role of Selected Mutations in the Q/N Rich Region of TDP-43 in EGFP-12xQ/N-Induced Aggregate Formation. Brain Res. 2012, 1462, 139–150. [Google Scholar] [CrossRef]
- Corcia, P.; Valdmanis, P.; Millecamps, S.; Lionnet, C.; Blasco, H.; Mouzat, K.; Daoud, H.; Belzil, V.; Morales, R.; Pageot, N.; et al. Phenotype and Genotype Analysis in Amyotrophic Lateral Sclerosis with TARDBP Gene Mutations. Neurology 2012, 78, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Wang, H.; Liu, J.; Wang, Z.; Xu, B.; Zhao, K.; Tao, X.; He, Z.; Yang, F.; Huang, X. Genetic and Clinical Features of Chinese Sporadic Amyotrophic Lateral Sclerosis Patients with TARDBP Mutations. Brain Behav. 2021, 11, e2312. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Ratti, A.; Gellera, C.; Buratti, E.; Castellotti, B.; Carlomagno, Y.; Ticozzi, N.; Mazzini, L.; Testa, L.; Taroni, F.; et al. High Frequency of TARDBP Gene Mutations in Italian Patients with Amyotrophic Lateral Sclerosis. Hum. Mutat. 2009, 30, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-J.; Lin, H.-X.; Liu, G.-L.; Tao, Q.-Q.; Ni, W.; Xiao, B.-G.; Wu, Z.-Y. The Investigation of Genetic and Clinical Features in Chinese Patients with Juvenile Amyotrophic Lateral Sclerosis. Clin. Genet. 2017, 92, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Newell, K.; Paron, F.; Mompean, M.; Murrell, J.; Salis, E.; Stuani, C.; Pattee, G.; Romano, M.; Laurents, D.; Ghetti, B.; et al. Dysregulation of TDP-43 Intracellular Localization and Early Onset ALS Are Associated with a TARDBP S375G Variant. Brain Pathol. 2019, 29, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Fu, S.; Lei, J.; Wu, H.; Shi, S.; Chen, K.; Hu, J.; Xu, X. Identification of Novel FUS and TARDBP Gene Mutations in Chinese Amyotrophic Lateral Sclerosis Patients with HRM Analysis. Aging 2020, 12, 22859. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Peng, Y.; Wang, X.-N.; Liu, M.-S.; Li, X.-G.; Cui, L.-Y. Screening of the TARDBP Gene in Familial and Sporadic Amyotrophic Lateral Sclerosis Patients of Chinese Origin. Neurobiol. Aging 2012, 33, 2229.e11–2229.e18. [Google Scholar] [CrossRef] [PubMed]
- Black, H.A.; Leighton, D.J.; Cleary, E.M.; Rose, E.; Stephenson, L.; Colville, S.; Ross, D.; Warner, J.; Porteous, M.; Gorrie, G.H.; et al. Genetic Epidemiology of Motor Neuron Disease-Associated Variants in the Scottish Population. Neurobiol. Aging 2017, 51, 178.e11–178.e20. [Google Scholar] [CrossRef]
- Kirby, J.; Goodall, E.F.; Smith, W.; Highley, J.R.; Masanzu, R.; Hartley, J.A.; Hibberd, R.; Hollinger, H.C.; Wharton, S.B.; Morrison, K.E.; et al. Broad Clinical Phenotypes Associated with TAR-DNA Binding Protein (TARDBP) Mutations in Amyotrophic Lateral Sclerosis. Neurogenetics 2010, 11, 217–225. [Google Scholar] [CrossRef]
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H. Amyotrophic Lateral Sclerosis Onset Is Influenced by the Burden of Rare Variants in Known Amyotrophic Lateral Sclerosis Genes: Rare Variants in ALS Genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef]
- Chen, W.; Xie, Y.; Zheng, M.; Lin, J.; Huang, P.; Pei, Z.; Yao, X. Clinical and Genetic Features of Patients with Amyotrophic Lateral Sclerosis in Southern China. Eur. J. Neurol. 2020, 27, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, R.-L.; Zhang, W.; Che, C.-H.; Feng, S.-Y.; Huang, H.-P.; Liu, C.-Y.; Zou, Z.-Y. Novel TARDBP Missense Mutation Caused Familial Amyotrophic Lateral Sclerosis with Frontotemporal Dementia and Parkinsonism. Neurobiol. Aging 2021, 107, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Min, J.; Staropoli, J.F.; Collin, E.; Bi, S.; Feng, X.; Barone, R.; Cao, Y.; O’Malley, L.; Xin, W.; et al. SOD1, ANG, TARDBP and FUS Mutations in Amyotrophic Lateral Sclerosis: A United States Clinical Testing Lab Experience. Amyotroph. Lateral Scler. 2012, 13, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Caroppo, P.; Camuzat, A.; Guillot-Noel, L.; Thomas-Antérion, C.; Couratier, P.; Wong, T.H.; Teichmann, M.; Golfier, V.; Auriacombe, S.; Belliard, S.; et al. Defining the Spectrum of Frontotemporal Dementias Associated with TARDBP Mutations. Neurol. Genet. 2016, 2, e80. [Google Scholar] [CrossRef]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Parish, L.D.; Occhineri, P.; Cau, T.B.; et al. Genetic Architecture of ALS in Sardinia. Neurobiol. Aging 2014, 35, 2882.e7–2882.e12. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome Sequencing in Amyotrophic Lateral Sclerosis Identifies Risk Genes and Pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Brenner, D.; Müller, K.; Wieland, T.; Weydt, P.; Böhm, S.; Lulé, D.; Hübers, A.; Neuwirth, C.; Weber, M.; Borck, G.; et al. NEK1 Mutations in Familial Amyotrophic Lateral Sclerosis. Brain 2016, 139, e28. [Google Scholar] [CrossRef]
- Kenna, K.P.; van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; van Rheenen, W.; van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 Variants Confer Susceptibility to Amyotrophic Lateral Sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Van Mossevelde, S.; Dillen, L.; De Bleecker, J.L.; Moisse, M.; Van Damme, P.; Van Broeckhoven, C.; van der Zee, J.; Engelborghs, S.; Crols, R.; et al. NEK1 Genetic Variability in a Belgian Cohort of ALS and ALS-FTD Patients. Neurobiol. Aging 2018, 61, 255.e1–255.e7. [Google Scholar] [CrossRef]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Yoshimura, J.; Doi, K.; Morishita, S.; Goto, J.; Toda, T.; et al. Loss-of-Function Variants in NEK1 Are Associated with an Increased Risk of Sporadic ALS in the Japanese Population. J. Hum. Genet. 2021, 66, 237–241. [Google Scholar] [CrossRef]
- Lattante, S.; Doronzio, P.N.; Conte, A.; Marangi, G.; Martello, F.; Bisogni, G.; Meleo, E.; Colavito, D.; Del Giudice, E.; Patanella, A.K.; et al. Novel Variants and Cellular Studies on Patients’ Primary Fibroblasts Support a Role for NEK1 Missense Variants in ALS Pathogenesis. Hum. Mol. Genet. 2021, 30, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; He, X.; Cui, B.; Zhao, F.; Zhou, C. NEK1 Mutations and the Risk of Amyotrophic Lateral Sclerosis (ALS): A Meta-Analysis. Neurol. Sci. 2021, 42, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).