
Complete Chloroplast Genome Features of Dendrocalamusfarinosus and Its Comparison and Evolutionary Analysis with Other Bambusoideae Species

Abstract
:
1. Introduction
2. Results and Discussion
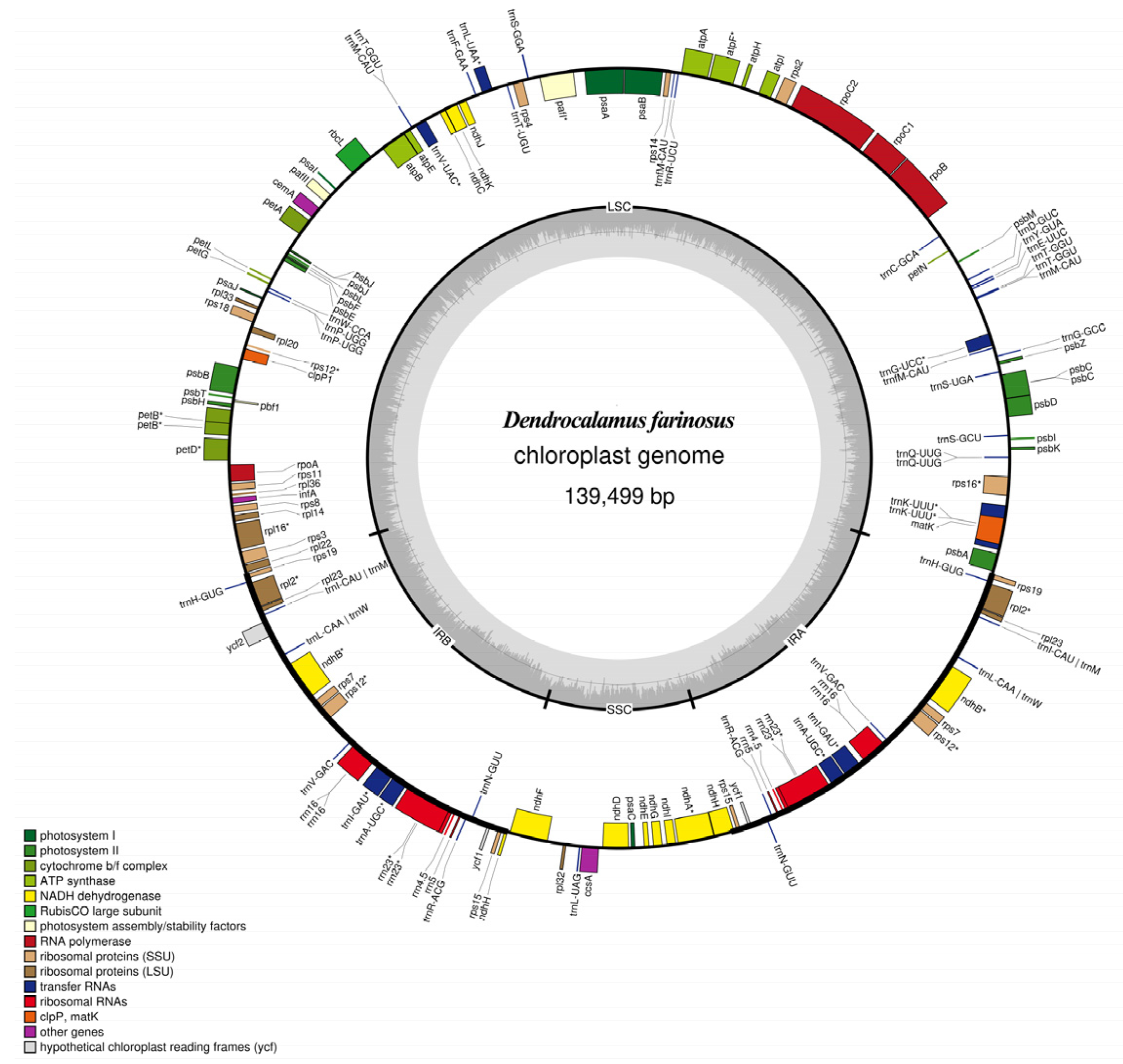
2.1. Characteristics of the D. farinosus cp Genome
2.2. Repeat Analyses
2.2.1. Long Repeats
2.2.2. Simple Sequence Repeats (SSRs)
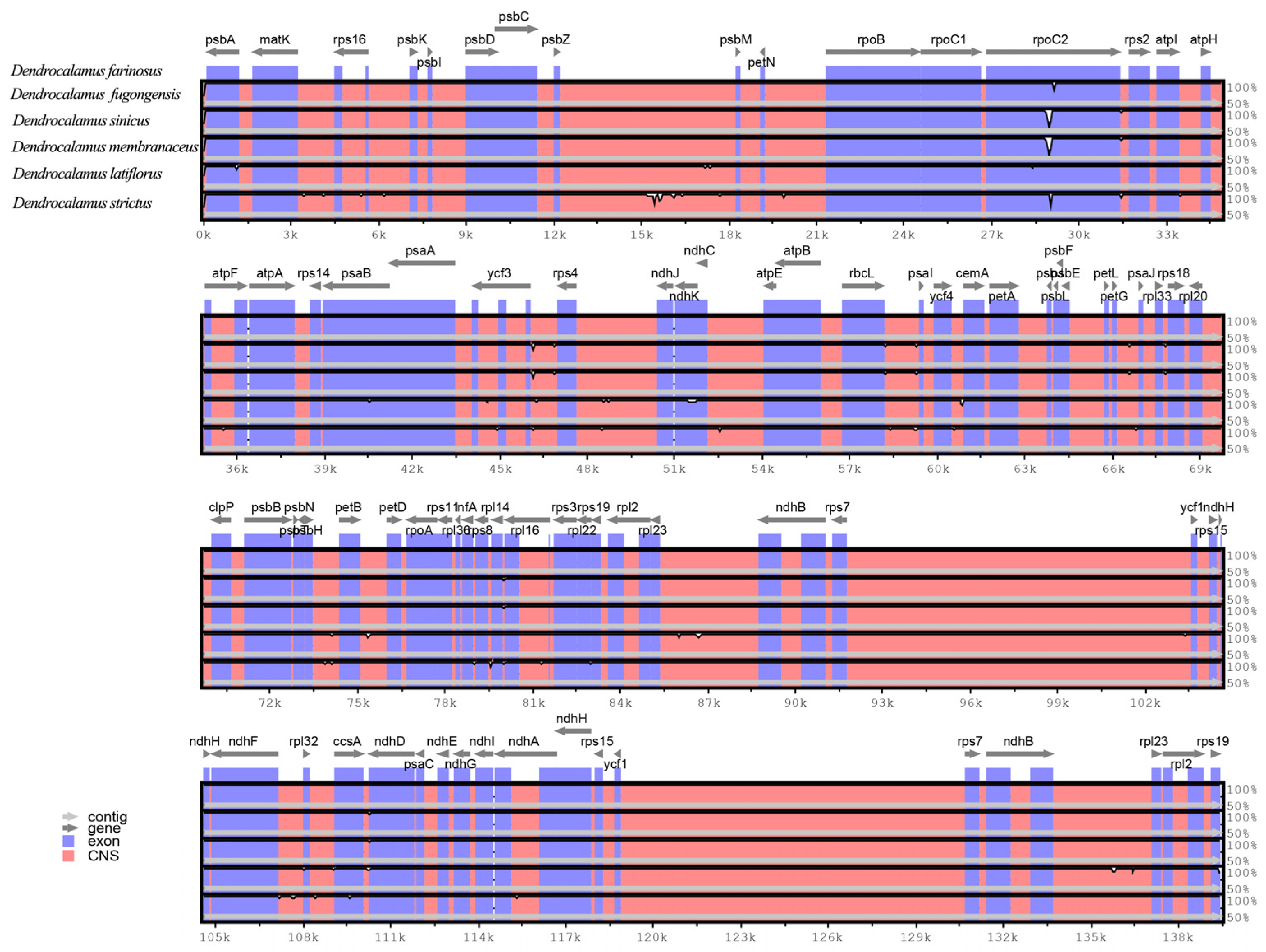
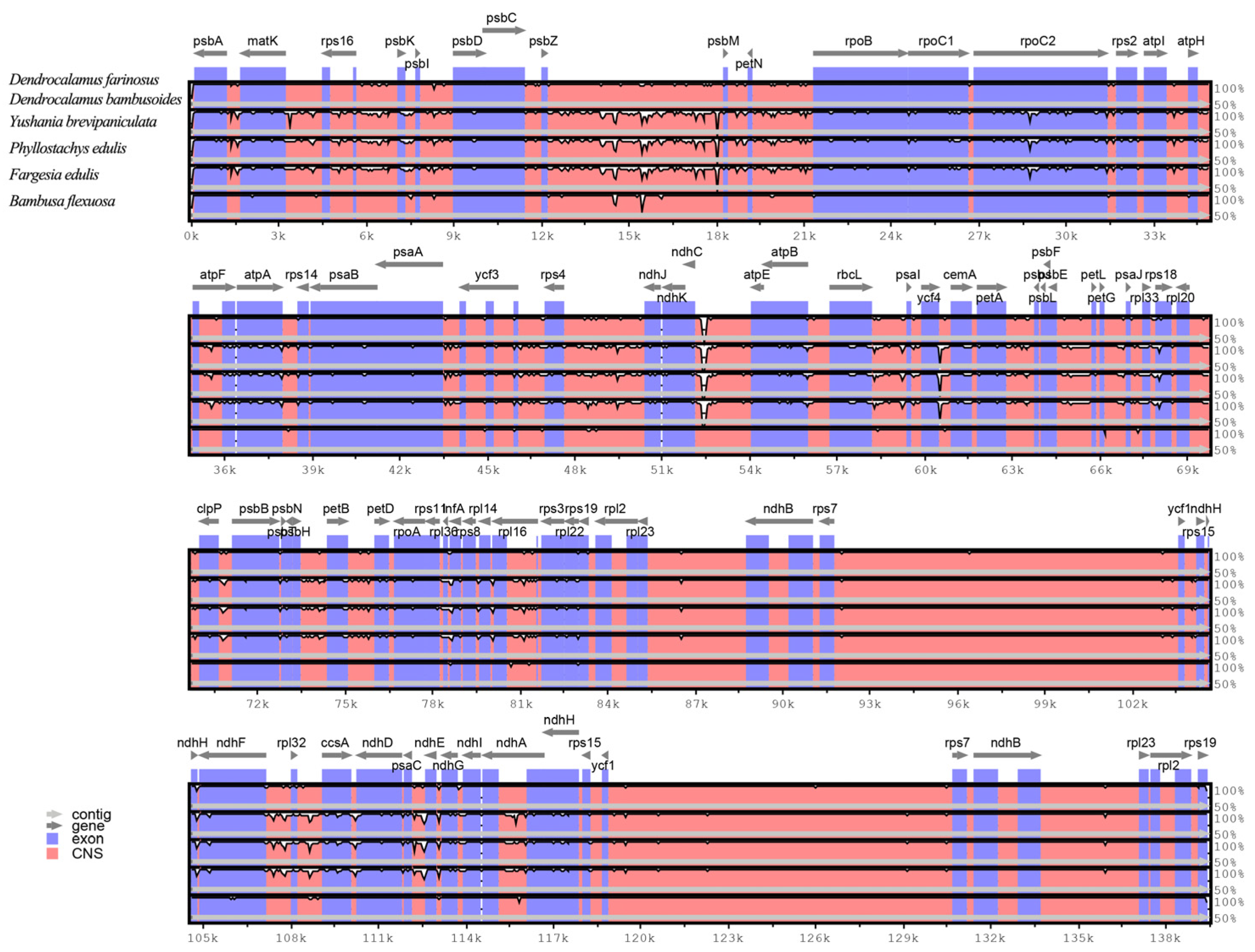
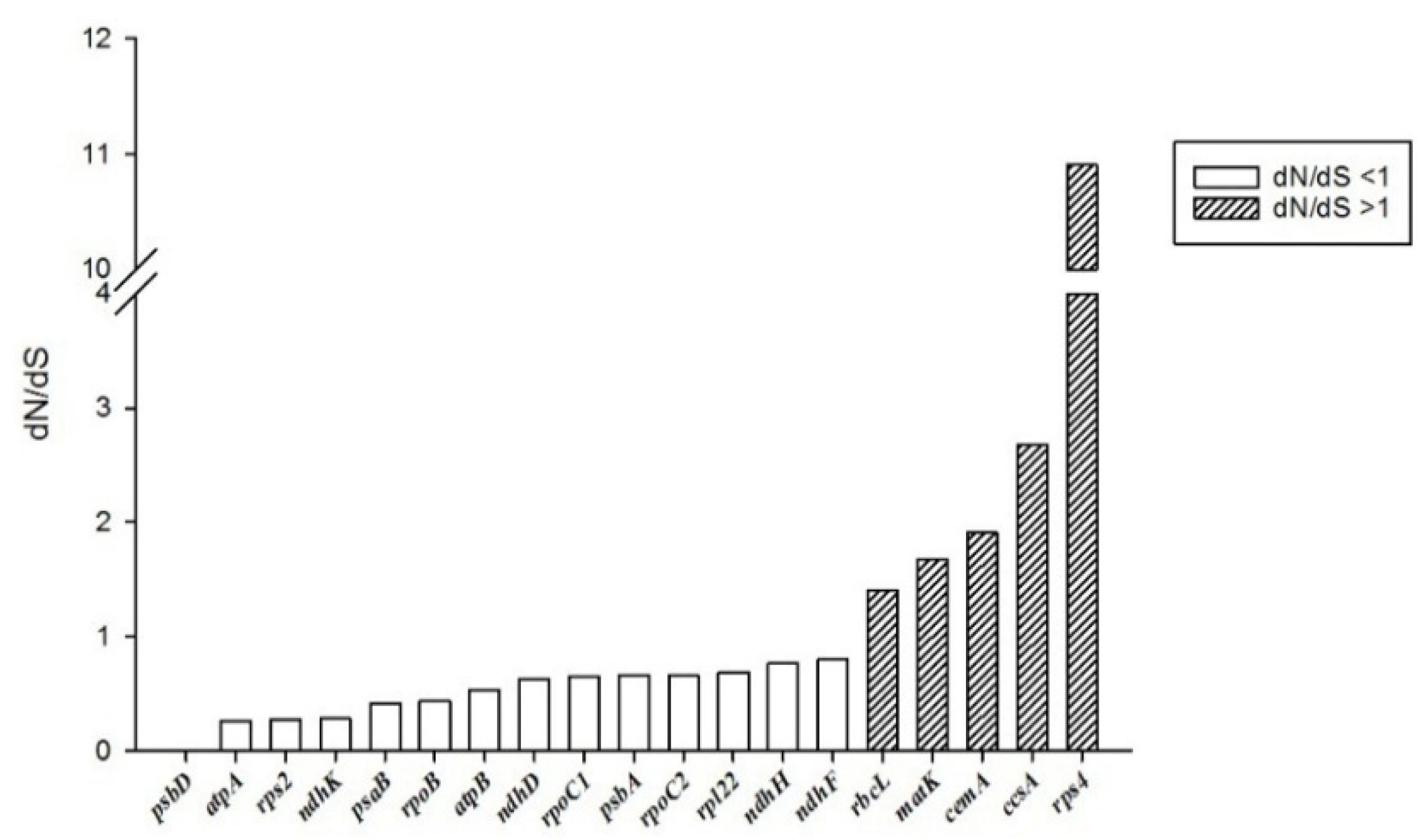
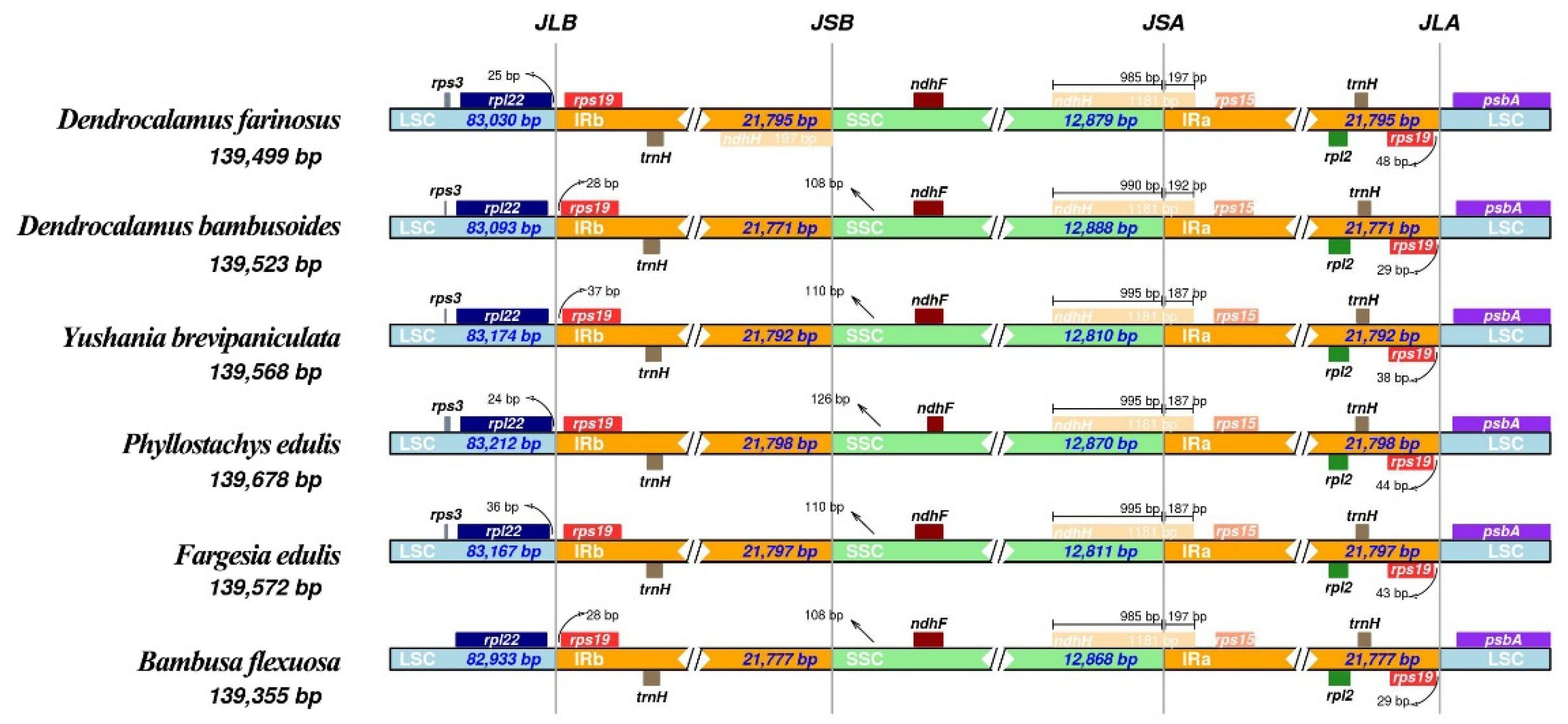
2.3. Comparative Analysis of Plastomes of Bambusoideae Species
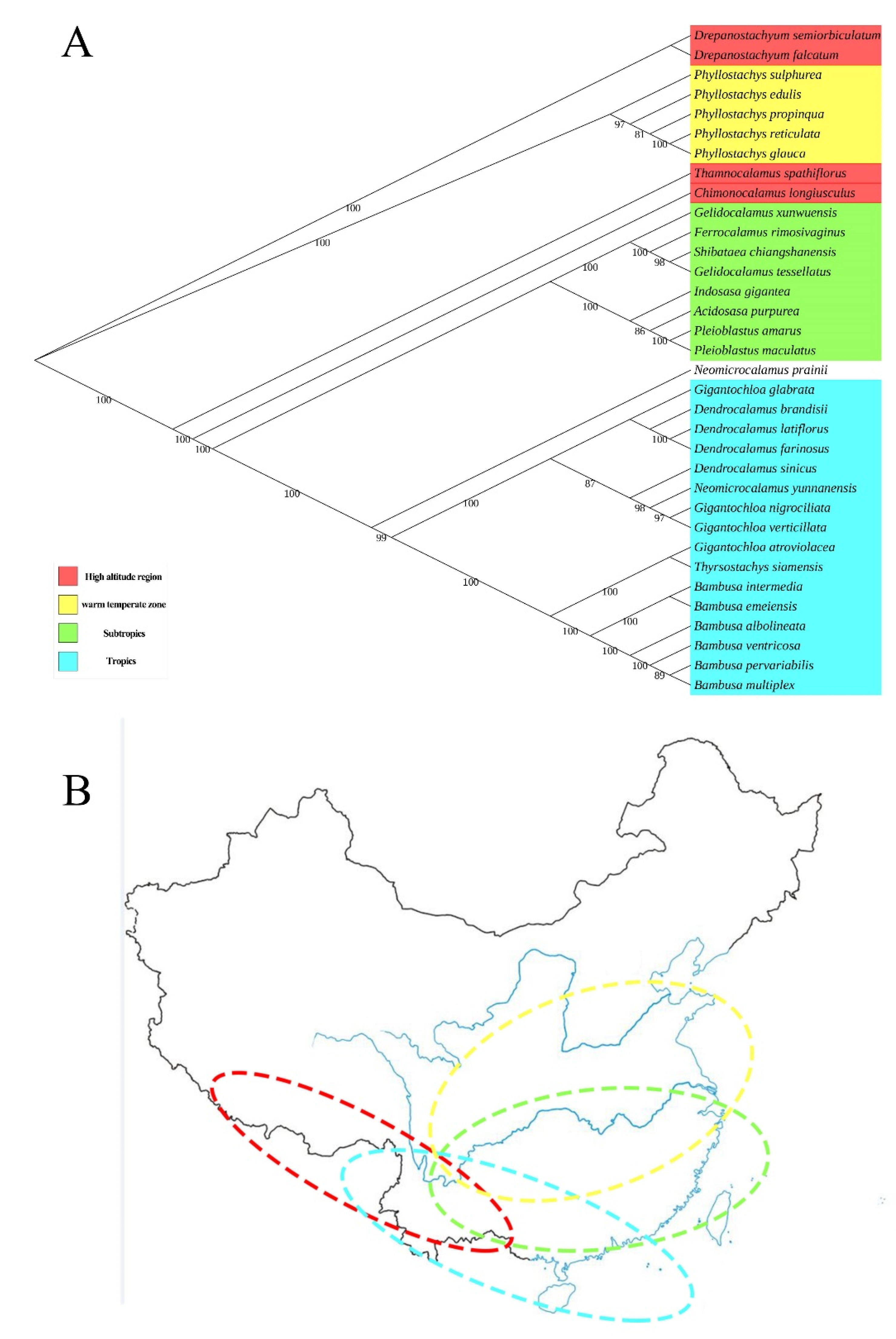
2.4. Phylogenetic Analysis
3. Materials and Methods
3.1. Plant Material and DNA Extraction
3.2. Chloroplast Genome Assembly and Gene Annotation
3.3. Repeat Analysis
3.4. Codon Bias Usage Analysis
3.5. Comparison of Related cp Genomes
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, J.X.; Zhou, M.Y.; Yang, G.Q.; Zhang, Y.X.; Ma, P.F.; Guo, C.; Li, D.Z. ddRAD analyses reveal a credible phylogenetic relationship of the four main genera of Bambusa-Dendrocalamus-Gigantochloa complex (Poaceae: Bambusoideae). Mol. Phylogenet Evol. 2020, 146, 106758. [Google Scholar] [CrossRef] [PubMed]
- Santosh, S.; Lalit, M.B.; Poonam, S.; Naik, S.N. Bamboo shoot processing: Food quality and safety aspect (a review). Trends Food Sci. Technol. 2010, 21, 181–189. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Zhang, Q.; Jiang, S. Present situation and the countermeasure analysis of bamboo timber processing industry in China. J. For. Eng. 2016, 1, 2–7. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K. Chlorophylls and carotenoids: Pigments of photosynthetic biomembranes. Meth. Enzymol. 1987, 148, 350–382. [Google Scholar] [CrossRef]
- Janzen, D.H. Why Bamboos Wait So Long to Flower. Annu. Rev. Ecol. Syst. 1976, 7, 347–391. [Google Scholar] [CrossRef]
- Yang, H.Q.; Xie, N.; Sun, M.S.; Xu, T.; Li, D.Z. Dendrocalamus atroviridis (Poaceae: Bambusoideae, Bambuseae), a new species from Southwest China. Phytotaxa 2016, 243, 170. [Google Scholar] [CrossRef]
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef]
- Fan, S.J.; Guo, X.X. Advances in research and application of plant chloroplast genome. J. Shandong Normal Univ. (Nat. Sci.) 2022, 37, 22–31. [Google Scholar] [CrossRef]
- Li, Y.H.; Ren, Y.K.; Zhao, X.H.; Liu, J.; Han, B.; Wang, C.B.; Tang, Z.H. Research Progress on Chloroplast Genome of Major Gramineous Crops. Biotechnol. Bull. 2020, 36, 112–121. [Google Scholar] [CrossRef]
- Howe, C.J. Chloroplast Genome. In eLS; John Wiley & Sons, Ltd: Chichester, UK, 2012. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef]
- Jansen, R.K.; Cai, Z.Q.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Mack, J.L.; Müller, K.F.; Bellian, M.G.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.H.; Kan, D.P.; Lee, S.B.; Daniell, H.; Lee, Y.W.; Lin, C.C.; Lin, C.S. Complete nucleotide sequence of Dendrocalamus latiflorus and Bambusa oldhamii chloroplast genomes. Tree Physiol. 2009, 6, 847. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.V.; Grennan, C.P.; Duvall, M.R. Plastome sequences of two New World bamboos—Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)--extend phylogenomic understanding of Bambusoideae. Am. J. Bot. 2012, 99, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.V.; Clark, L.G.; Triplett, K.J.; Grennan, C.P.; Duvall, M.R. Biogeography and phylogenomics of New World Bambusoideae (Poaceae), revisited. Am. J. Bot. 2014, 101, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, W.P.; Clark, L.G.; Attigala, L.; Sanchez, E.R.; Duvall, M.R. Evolution of the bamboos (Bambusoideae; Poaceae): A full plastome phylogenomic analysis. BMC Evol. Biol. 2015, 15, 50. [Google Scholar] [CrossRef]
- Chen, P.; Zheng, R.H.; Wang, Y.; Hu, Y.B. Study on clonal selection of Dendrocalamus farinosus in south Sichuan. J. Sichuan For. Sci. Technol. 2021, 42, 109–113. [Google Scholar] [CrossRef]
- Fu, M.D.W. Research Progress in Overview of Bio-ecological Characteristics and Cultivation Technology for Dendrocalamus farinosus. World Bamboo Ratt. 2014, 12, 25–28. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, X.; Li, Y.; Zhang, J. The complete chloroplast genome sequence of Dendrocalamus sinicus. Mitochondrial DNA B Resour. 2019, 4, 2988–2989. [Google Scholar] [CrossRef]
- Tang, S.L.; Xie, J.H.; Cai, J.Z. The complete plastid genome of Thyrsostachys siamensis (Poaceae, Bambusoideae). Mitochondrial DNA B Resour. 2021, 6, 1781–1783. [Google Scholar] [CrossRef]
- Ya-Ping, H.; Jie, Z.; Zhao-Yan, Y.; Jia-Jia, L.; Ming-Ye, X.; Qi-Rong, G. The complete chloroplast genome of Phyllostachys heteroclada f. solida (Poaceae). Mitochondrial DNA B Resour. 2021, 6, 566–567. [Google Scholar] [CrossRef]
- Jie, Z.; Yaping, H.; Zhaoyan, Y.; Jiajia, L.; Mingye, X.; Qirong, G. The complete chloroplast genome of a solid type of Phyllostachys nidularia (Bambusoideae: Poaceae), a species endemic to China. Mitochondrial DNA B Resour. 2021, 6, 978–979. [Google Scholar] [CrossRef]
- Hecht, A.; Glasgow, J.; Jaschke, P.R.; Bawazer, L.A.; Munson, M.S.; Cochran, J.R.; Salit, M. Measurements of translation initiation from all 64 codons in E. coli. Nucleic Acids Res. 2017, 45, 3615–3626. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, D.A. Complete Chloroplast Genome of Abutilon fruticosum: Genome Structure, Comparative and Phylogenetic Analysis. Plants 2021, 10, 270. [Google Scholar] [CrossRef]
- Gu, C.; Tembrock, L.R.; Zheng, S.; Wu, Z. The Complete Chloroplast Genome of Catha edulis: A Comparative Analysis of Genome Features with Related Species. Int. J. Mol. Sci. 2018, 19, 525. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Qian, F.; Hou, Y.; Yang, W.; Cai, M.; Wu, X. Complete chloroplast genome features and phylogenetic analysis of Eruca sativa (Brassicaceae). PLoS ONE 2021, 16, e0248556. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.; Shanker, A. Identification of Simple Sequence Repeats in Chloroplast Genomes of Magnoliids Through Bioinformatics Approach. Interdiscip Sci. 2016, 8, 327–336. [Google Scholar] [CrossRef]
- Du, X.; Zeng, T.; Feng, Q.; Hu, L.; Luo, X.; Weng, Q.; Zhu, B. The complete chloroplast genome sequence of yellow mustard (Sinapis alba L.) and its phylogenetic relationship to other Brassicaceae species. Gene 2020, 731, 144340. [Google Scholar] [CrossRef]
- Yan, C.; Du, J.; Gao, L.; Li, Y.; Hou, X. The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 2019, 699, 24–36. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Ma, P.F.; Li, D.Z. High-Throughput Sequencing of Six Bamboo Chloroplast Genomes: Phylogenetic Implications for Temperate Woody Bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.J.; Lee, H.L. Complete Chloroplast Genome Sequences from Korean Ginseng (panax schinseng nees) and Comparative Analysis of Sequence Evolution among 17 Vascular Plants. DNA Res. 2004, 4, 247–261. [Google Scholar] [CrossRef]
- Tong, W.; Kim, T.S.; Park, Y.J. Rice Chloroplast Genome Variation Architecture and Phylogenetic Dissection in Diverse Oryza Species Assessed by Whole-Genome Resequencing. Rice 2016, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.P.; Bi, Y.; Yang, F.P.; Zhang, M.F.; Chen, X.Q.; Xue, J.; Zhang, X.H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Hou, D.; Zhuo, J.; Zheng, R.; Wang, Y.; Li, B.; Lin, X. Complete chloroplast genome sequence of Bambusa rigida (Bambuseae). Mitochondrial DNA B Resour. 2020, 5, 2972–2973. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.L.; Chandran, S.; Franklin, D.C.; Isagi, Y.; Koshy, K.C.; Sungkaew, S.; Wong, K.M. Multi-gene Region Phylogenetic Analyses Suggest Reticulate Evolution and a Clade of Australian Origin Among Paleotropical Woody Bamboos (poaceae: Bambusoideae: Bambuseae). Plant Syst. Evol. 2013, 299, 239–257. [Google Scholar] [CrossRef]
- Wen, T.H. Some ideas about the origin of bamboos. J. Bamboo Res. 1983, 2, 1–10. [Google Scholar]
- Wu, Z.Y. Flora of China; Science Press: Beijing, China, 2013. [Google Scholar]
- Hanjiao, G.; Cancan, Z.; Jinsong, W.; Xuewen, S.; Ruixue, X.; Bin, L.; Fusheng, C.; Wensheng, B. Variation in basic morphological and functional traits of Chinese bamboo. Biodivers. Sci. 2019, 27, 10. [Google Scholar] [CrossRef]
- Doyle, J. Dna Protocols for Plants. In Molecular Techniques in Taxonomy; Hewitt, G.M., Johnston, A.W.B., Young, J.P.W., Eds.; Springer: Berlin, Germany, 1991; Volume 57, pp. 283–293. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hughes, A.L.; Bano, N.; McArdle, S.; Livingston, S.; Deubner, H.; Gretch, D.R. Genetic diversity of near genome-wide hepatitis C virus sequences during chronic infection: Evidence for protein structural conservation over time. PLoS ONE 2011, 6, e19562. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Group of Genes | Name of Genes |
|---|---|---|
| Genes for photosynthesis | Subunits of ATP synthase | atpA, atpB, atpE, atpF+, atpH, atpI |
| Subunits of NADH-dehydrogenase | ndhA +, ndhB +,a, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB +, petD+, petG, petL, petN | |
| Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Subunits of photosystem II | psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbT, psbZ | |
| Subunit of rubisco | rbcL | |
| photosystem assembly/stability factors | pafI ++, pafII, pbf1 | |
| Self replication | Large subunit of ribosome | rpl2+,a, rpl14, rpl16, rpl20, rpl22, rpl23 a, rpl32, rpl33, rpl36 |
| Small subunit of ribosome | rps2, rps3, rps4, rps7a, rps8, rps11, rps12++,a, rps12-fragment+, rps14, rps15a, rps16 +, rps18, rps19 a | |
| DNA dependent RNA polymerase | rpoA, rpoB, rpoC1+, rpoC2 | |
| tRNA Genes | trnA-UGC+,a, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC +, trnH-GUGa, trnI-CAUa, trnI-GAU +,a, trnK-UUU +, trnL-CAA a, trnL-UAA +, trnL-UAG, trnM-CAU, trnN-GUU a, trnP-UGG, trnQ-UUG, trnR-ACGa, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC a, trnV-UAC+, trnW-CCA, trnY-GUA | |
| rRNA Genes | rrn4.5a, rrn5a, rrn16 a, rrn23+,a | |
| Other genes | c-type cytochrom synthesis gene | ccsA |
| Envelop membrane protein | cemA | |
| Protease | clpP1 | |
| Translational initiation factor | infA | |
| Maturase | matK | |
| Component of TIC complex | ycf2, ycf15 a | |
| Subunit acetyl-coA carboxylase | accD |
| S. No | Gene | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|---|
| 1 | rps16 | LSC | 36 | 846 | 222 | ||
| 2 | atpF | LSC | 158 | 837 | 409 | ||
| 3 | pafI | LSC | 132 | 732 | 226 | 724 | 161 |
| 4 | rpl2 | IR | 405 | 660 | 432 | ||
| 5 | ndhB | IR | 777 | 712 | 756 | ||
| 6 | rps12 | IR | 232 | 537 | 36 | ||
| 7 | ndhA | SSC | 548 | 1026 | 541 | ||
| 8 | trnK-UUU | LSC | 38 | 2505 | 34 | ||
| 9 | trnG-UCC | LSC | 23 | 673 | 48 | ||
| 10 | trnL-UAA | LSC | 35 | 539 | 50 | ||
| 11 | trnV-UAC | LSC | 39 | 596 | 37 | ||
| 12 | trnI-GAU | IR | 42 | 946 | 35 | ||
| 13 | trnA-UGC | IR | 38 | 811 | 35 |
| SSR Type | Unit | Length | Number | Position on Genome (bp) |
|---|---|---|---|---|
| P1 | A | 10 | 4 | 7647–7656, 12,227–12,236, 31,304–31,313, 39,008–39,017 |
| A | 11 | 3 | 49,902–49,912, 107,247–107,257, 139,481–139,491 | |
| A | 12 | 2 | 45,950–45,961, 46,921–46,932 | |
| A | 13 | 1 | 110,286–110,298 | |
| T | 10 | 4 | 38,212–38,221, 43,628–43,637, 67,161–67,170, 82,514–82,523 | |
| T | 11 | 2 | 34,229–34,239, 79,557–79,567 | |
| T | 12 | 1 | 51,765–51,776 | |
| P2 | AT | 10 | 1 | 27,587–27,596 |
| TA | 10 | 2 | 87,344–87,353, 135,177–135,186 | |
| TC | 10 | 1 | 117,486–117,495 | |
| p3 | AAT | 12 | 1 | 26,292–26,303 |
| CAG | 12 | 1 | 684–695 | |
| TTC | 12 | 1 | 67,459–67,470 | |
| P4 | AAAG | 12 | 1 | 18,988–18,999 |
| AACG | 12 | 1 | 101,658–101,669 | |
| AATA | 12 | 1 | 110,503–110,514 | |
| ATAC | 12 | 1 | 17,899–17,910 | |
| GAAA | 12 | 1 | 65,129–65,140 | |
| GTAG | 16 | 1 | 53,971–53,986 | |
| TATT | 12 | 1 | 50,161–50,172 | |
| TCCT | 12 | 1 | 44,873–44,884 | |
| TCGT | 12 | 1 | 120,860–120,871 | |
| p5 | TTTTA | 15 | 1 | 31,515–31,529 |
| C | (A)10/(A)11 | 50 | 1 | 35,536–35,585 |
| (T)10/(T)10 | 28 | 1 | 78,948–78,975 | |
| (T)10/(T)10 | 32 | 1 | 8601–8632 | |
| (T)10/(A)13 | 106 | 1 | 48,475–48,580 | |
| (AGAA)3/(T)11 | 39 | 1 | 70,751–70,789 | |
| (TTTA)3/(A)13 | 61 | 1 | 74,099–74,159 | |
| (TCT)4/(T)11 | 95 | 1 | 82,955–83,049 | |
| (T)10/(AATA)3/(TATT)3 | 143 | 1 | 58,342–58,484 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, J.; Wang, Y.; Zhuo, J.; Gao, H.; Vasupalli, N.; Hou, D.; Lin, X. Complete Chloroplast Genome Features of Dendrocalamusfarinosus and Its Comparison and Evolutionary Analysis with Other Bambusoideae Species. Genes 2022, 13, 1519. https://doi.org/10.3390/genes13091519
Pei J, Wang Y, Zhuo J, Gao H, Vasupalli N, Hou D, Lin X. Complete Chloroplast Genome Features of Dendrocalamusfarinosus and Its Comparison and Evolutionary Analysis with Other Bambusoideae Species. Genes. 2022; 13(9):1519. https://doi.org/10.3390/genes13091519
Chicago/Turabian StylePei, Jialong, Yong Wang, Juan Zhuo, Huibin Gao, Naresh Vasupalli, Dan Hou, and Xinchun Lin. 2022. "Complete Chloroplast Genome Features of Dendrocalamusfarinosus and Its Comparison and Evolutionary Analysis with Other Bambusoideae Species" Genes 13, no. 9: 1519. https://doi.org/10.3390/genes13091519