
Changes in the Genetic Structure of Lithuania’s Wild Boar (Sus scrofa) Population Following the Outbreak of African Swine Fever

Abstract
:1. Introduction
2. Materials and Methods
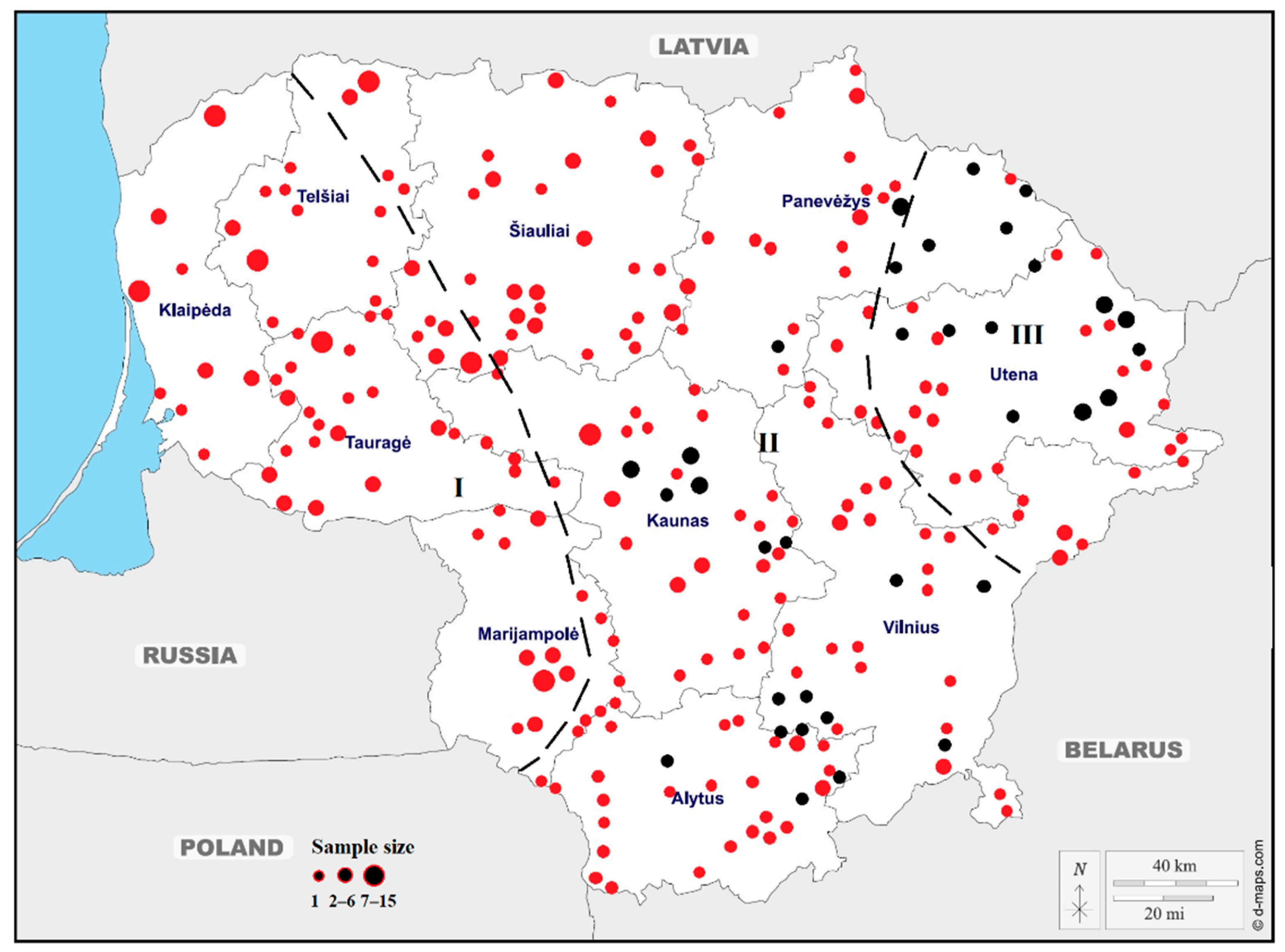
2.1. Population Sampling and Genomic DNA Extraction
2.2. Microsatellite Analysis
2.3. Data Analysis
3. Results
3.1. SSR Genetic Diversity and Polymorphism
3.2. Null Alleles
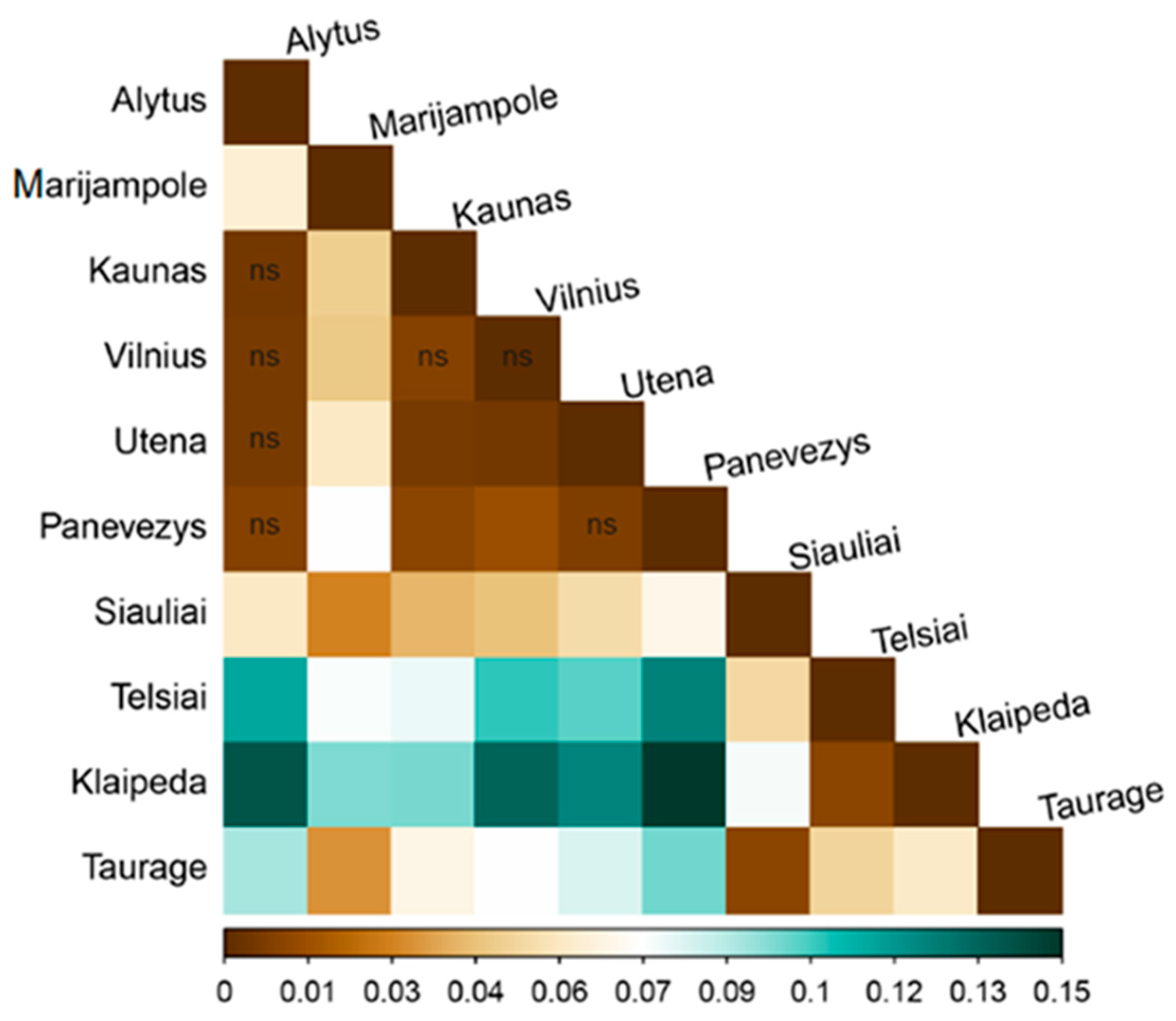
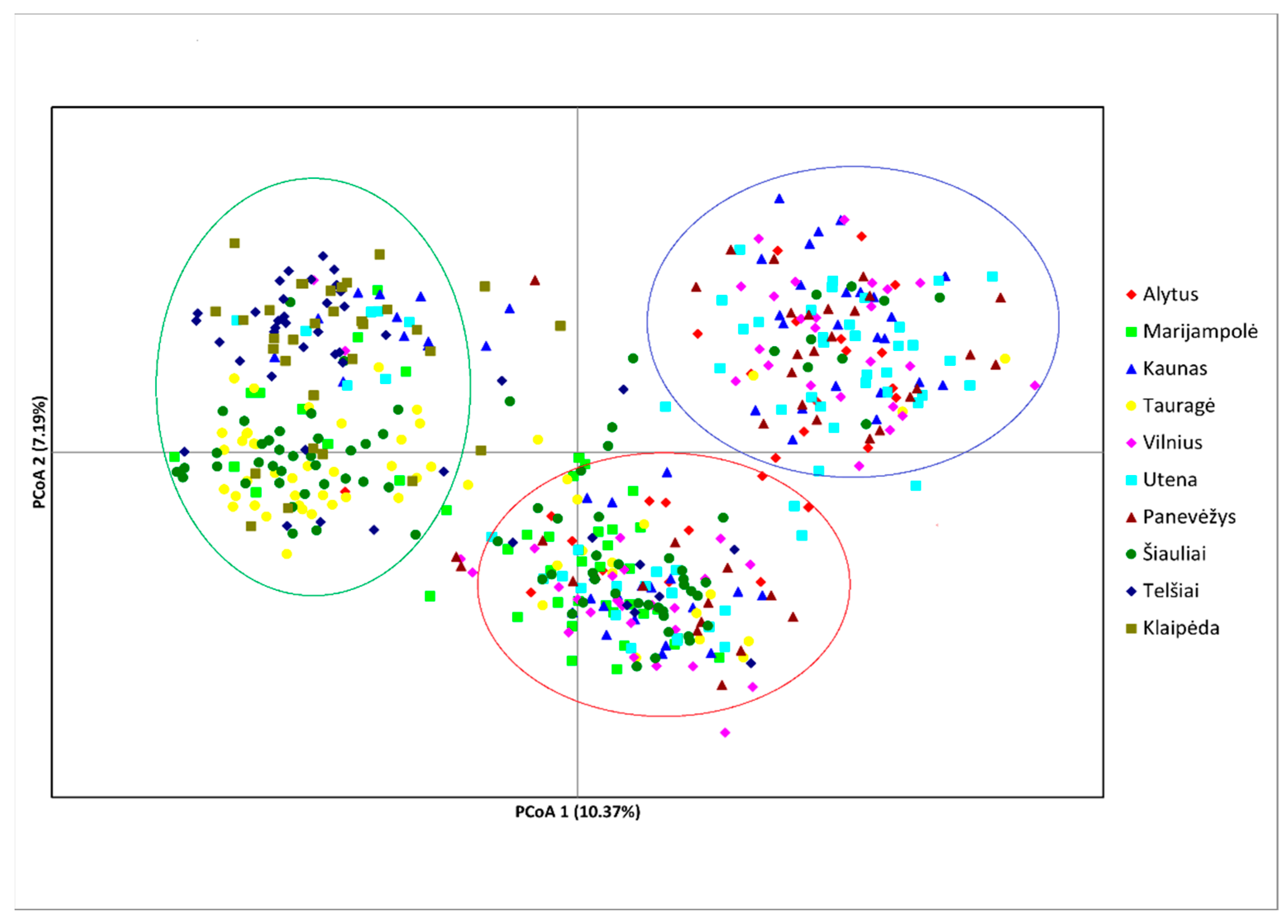
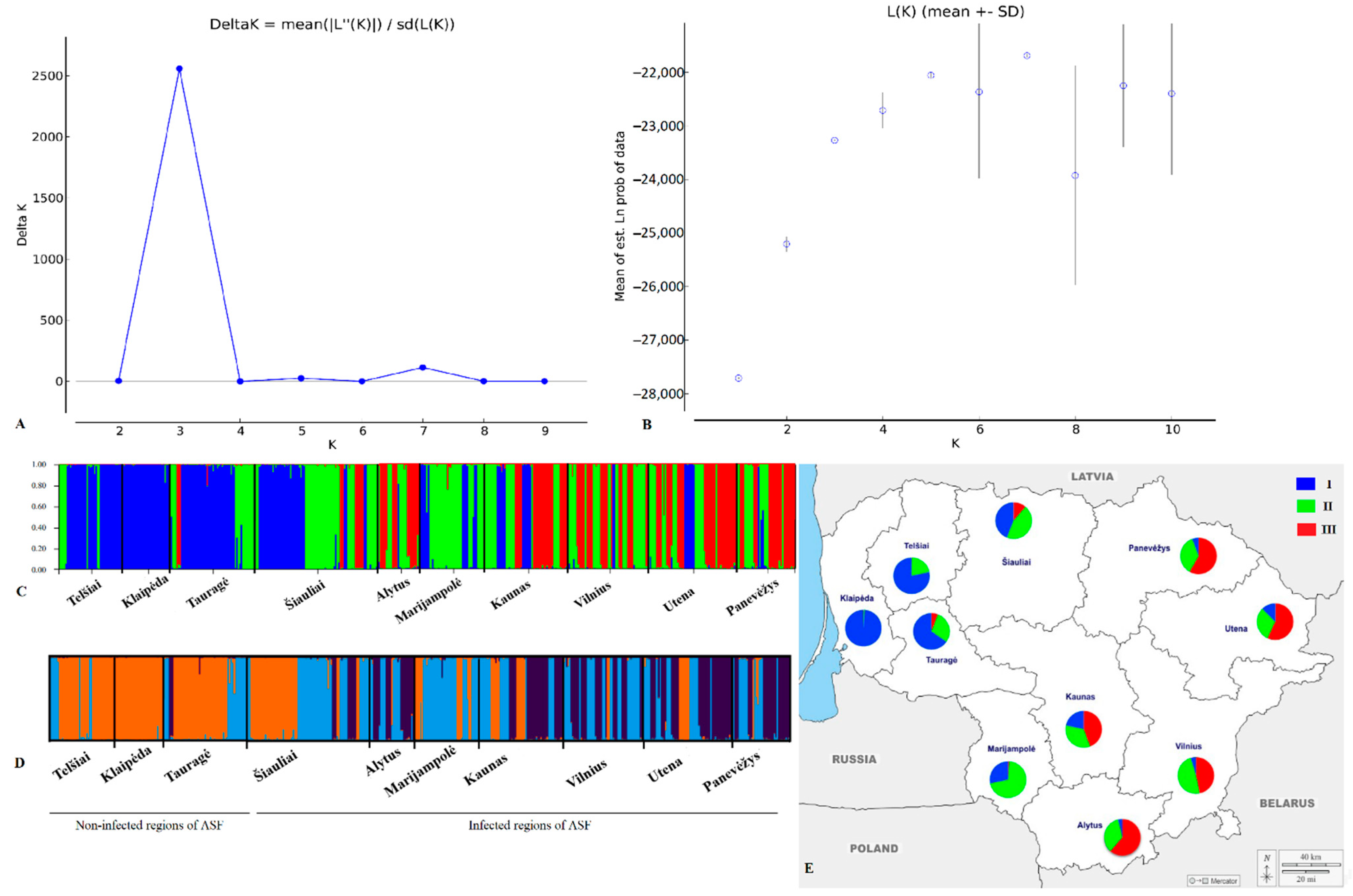
3.3. Population Differentiation, Genetic Distance and Genetic Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Apollonio, M.; Andersen, R.; Putman, R. European Ungulates and Their Management in the 21st Century; Cambridge University Press: Cambridge, UK, 2010; p. 604. [Google Scholar]
- Barrios-Garcia, M.N.; Ballari, S.A. Impact of wild boar (Sus scrofa) in its introduced and native range: A review. Biol. Invasions. 2012, 14, 2283–2300. [Google Scholar] [CrossRef]
- Massei, G.; Kindberg, J.; Licoppe, A.; Gačić, D.; Šprem, N.; Kamler, J.; Baubet, E.; Hohmann, U.; Monaco, A.; Ozoliņš, J.; et al. Wild boar populations up, numbers of hunters down? A review of trends and implications for Europe. Pest. Manag. Sci. 2015, 71, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Vetter, S.G.; Ruf, T.; Bieber, C.; Arnold, W. What Is a Mild Winter? Regional Differences in Within-Species Responses to Climate Change. PLoS ONE 2015, 10, e0132178. [Google Scholar]
- Vetter, S.G.; Puskas, Z.; Bieber, C.; Ruf, T. How climate change and wildlife management affect population structure in wild boars. Sci. Rep. 2020, 10, 7298. [Google Scholar] [CrossRef] [PubMed]
- Pittiglio, C.; Khomenko, S.; Beltran-Alcrudo, D. Wild boar mapping using population-density statistics: From polygons to high resolution raster maps. PLoS ONE 2018, 13, e0193295. [Google Scholar] [CrossRef]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African swine fever virus isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority); Boklund, A.; Cay, B.; Depner, K.; Földi, Z.; Guberti, V.; Masiulis, M.; Miteva, A.; More, S.; Olsevskis, E.; et al. Epidemiological analyses of African swine fever in the European Union (November 2017 until November 2018). EFSA J. 2018, 16, 34–52. [Google Scholar]
- EFSA AHAW Panel (EFSA Panel on Animal Health and Welfare). Scientific opinion on African swine fever. EFSA J. 2015, 13, 4163–4192. [Google Scholar]
- Morelle, K.; Bubnicki, J.; Churski, M.; Gryz, J.; Podgórski, T.; Kuijper, D. Disease-Induced Mortality Outweighs Hunting in Causing Wild Boar Population Crash After African Swine Fever Outbreak. Front. Vet. Sci. 2020, 7, 378. [Google Scholar] [CrossRef]
- Mačiulskis, P.; Masiulis, M.; Pridotkas, G.; Buitkuvienė, J.; Jurgelevičius, V.; Jacevičienė, I.; Zagrabskaitė, R.; Zani, L.; Pilevičienė, S. The African Swine Fever Epidemic in Wild Boar (Sus scrofa) in Lithuania (2014–2018). Vet. Sci. 2020, 7, 15. [Google Scholar] [CrossRef]
- Alexandri, P.; Triantafyllidis, A.; Papakostas, S.; Chatzinikos, E.; Platis, P.; Papageorgiou, M.; Larson, G.; Abatzopoulos, J.T.; Triantaphyllidis, C. The Balkans and the colonization of Europe: The post-glacial range expansion of the wild boar, Sus scrofa. J. Biogeogr. 2012, 39, 713–723. [Google Scholar] [CrossRef]
- Kusza, S.; Podgórski, T.; Scandura, M.; Borowik, T.; Jávor, A.; Sidorovich, V.E.; Bunevich, A.N.; Kolesnikov, M.; Jędrzejewska, B. Contemporary genetic structure, phylogeography and past demographic processes of wild boar Sus scrofa population in Central and Eastern Europe. PLoS ONE 2014, 9, e91401. [Google Scholar] [CrossRef] [PubMed]
- Vilaca, S.T.; Biosa, D.; Zachos, F.; Iacolina, L.; Kirschning, J.; Alves, P.C. Mitochondrial phylogeography of the European wild boar: The effect of climate on genetic diversity and spatial lineage sorting across Europe. J. Biogeogr. 2014, 41, 987–998. [Google Scholar] [CrossRef]
- Khederzadeh, S.; Kusza, S.; Huang, C.P.; Markov, N.; Scandura, M.; Babaev, E.; Šprem, N.; Seryodkin, I.V.; Paule, L.; Esmailizadeh, A.; et al. Maternal genomic variability of the wild boar (Sus scrofa) reveals the uniqueness of East-Caucasian and Central Italian populations. Ecol. Evol. 2019, 9, 9467–9478. [Google Scholar] [CrossRef] [PubMed]
- Niedziałkowska, M.; Tarnowska, E.; Ligmanowska, J.; Jędrzejewska, B.; Podgórski, T.; Radziszewska, A.; Ratajczyk, I.; Kusza, S.; Bunevich, A.N.; Danila, G.; et al. Clear phylogeographic pattern and genetic structure of wild boar Sus scrofa population in Central and Eastern Europe. Sci. Rep. 2021, 11, 9680. [Google Scholar] [CrossRef] [PubMed]
- Vernesi, C.; Crestanello, B.; Pecchioli, E.; Tartari, D.; Caramelli, D.; Hauffe, H.; Bertorelle, G. The genetic impact of demographic decline and reintroduction in the wild boar (Sus scrofa): A microsatellite analysis. Mol. Ecol. 2003, 12, 585–595. [Google Scholar] [CrossRef]
- Nikolov, I.S.; Gum, B.; Markov, G.; Kuehn, R. Population genetic structure of wild boar Sus scrofa in Bulgaria as revealed by microsatellite analysis. Acta Theriol. 2009, 54, 193–205. [Google Scholar] [CrossRef]
- Ferreira, E.; Souto, L.; Soares, A.M.V.M.; Fonseca, C. Genetic structure of the wild boar population in Portugal: Evidence of a recent bottleneck. Mamm. Biol. 2009, 74, 274–285. [Google Scholar] [CrossRef]
- Veličković, N.; Ferreira, E.; Djan, M.; Ernst, M.; Obreht Vidaković, D.; Monaco, A.; Fonseca, C. Demographic history, current expansion and future management challenges of wild boar populations in the Balkans and Europe. Heredity 2016, 117, 348–357. [Google Scholar] [CrossRef]
- Alexandri, P.; Megens, H.J.; Crooijmans, R.P.M.A.; Groenen, M.A.M.; Goedbloed, D.J.; Herrero-Medrano, J.M.; Rund, L.A.; Schook, L.B.; Chatzinikos, E.; Triantaphyllidis, C.; et al. Distinguishing migration events of different timing for wild boar in the Balkans. J. Biogeogr. 2017, 44, 259–270. [Google Scholar] [CrossRef]
- Tajchman, K.; Drozd, L.; Karpiński, M.; Czyżowski, P.; Goleman, M. Population Genetic Structure of Wild Boars in Poland. Russ. J. Genet. 2018, 54, 548–553. [Google Scholar] [CrossRef]
- Mihalik, B.; Frank, K.; Astuti, P.K.; Szemethy, D.; Szendrei, L.; Szemethy, L.; Kusza, S.; Stéger, V. Population Genetic Structure of the Wild Boar (Sus scrofa) in the Carpathian Basin. Genes 2020, 11, 1194. [Google Scholar] [CrossRef]
- Griciuvienė, L.; Janeliūnas, Ž.; Jurgelevičius, V.; Paulauskas, A. The effects of habitat fragmentation on the genetic structure of wild boar (Sus scrofa) population in Lithuania. BMC Genom Data 2021, 22, 53. [Google Scholar] [CrossRef] [PubMed]
- Kupferschmid, F.A.L.; Crovadore, J.; Fischer, C.; Lefort, F. Shall the Wild Boar Pass? A Genetically Assessed Ecological Corridor in the Geneva Region. Sustainability 2022, 14, 7463. [Google Scholar] [CrossRef]
- FAO. Molecular Genetic Characterization of Animal Genetic Resources. FAO Animal Production and Health Guidelines. 2011, Volume 9, p. 85. Available online: https://www.fao.org/3/i2413e/i2413e00.pdf (accessed on 29 March 2022).
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes. 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices. Version 2.9.3. J. Hered. 2001, 86, 485–486. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. Genepop (Version-1.2) population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef]
- Oosterhout, C.V.; Hutchinson, W.F.; Wills, D.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes. 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Brookfield, J.F. A simple new method for estimating null allele frequency from heterozygote deficiency. Mol. Ecol. 1996, 5, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Šprem, N.; Safner, T.; Treer, T.; Florijančić, T.; Jurić, J.; Cubric-Curik, V.; Frantz, A.C.; Curik, I. Are the dinaric mountains a boundary between continental and mediterranean wild boar populations in Croatia? Eur. J. Wildl. Res. 2016, 62, 167–177. [Google Scholar] [CrossRef]
- Ishikawa, K.; Doneva, R.; Raichev, E.G.; Peeva, S.; Doichev, V.D.; Amaike, Y.; Nishita, Y.; Kaneko, Y.; Masuda, R. Population genetic structure and diversity of the East Balkan Swine (Sus scrofa) in Bulgaria, revealed by mitochondrial DNA and microsatellite analyses. Anim. Sci. J. 2021, 92, e13630. [Google Scholar] [CrossRef]
- Abramovs, N.; Brass, A.; Tassabehji, M. Hardy-Weinberg Equilibrium in the Large Scale Genomic Sequencing Era. Front. Genet. 2020, 11, 210. [Google Scholar] [CrossRef]
- Pautienius, A.; Grigas, J.; Pileviciene, S.; Zagrabskaite, R.; Buitkuviene, J.; Pridotkas, G.; Stankevicius, R.; Streimikyte, Z.; Salomskas, A.; Zienius, D.; et al. Prevalence and spatiotemporal distribution of African swine fever in Lithuania, 2014-2017. Virol. J. 2018, 15, 177. [Google Scholar] [CrossRef]
- European Commission (EC). Working Document Strategic Approach to the Management of African Swine Fever for the EU; Vol. SANTE/7113/2015—Rev 10; EC: Brussels, Belgium, 2018.
- Allendorf, F.W.; Hard, J.J. Human-induced evolution caused by unnatural selection through harvest of wild animals. PNAS 2009, 106, 9987–9994. [Google Scholar] [CrossRef]
- Harris, R.B.; Wall, W.A.; Allendorf, F.W. Genetic consequences of hunting: What do we know and what should we do? Wildl. Soc. Bull. 2002, 30, 634–643. [Google Scholar]
- Chapuis, M.P.; Estoup, A. Microsatellite Null Alleles and Estimation of Population Differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Rico, C.; Cuesta, J.A.; Drake, P.; Macpherson, E.; Bernatchez, L.; Marie, A.D. Null alleles are ubiquitous at microsatellite loci in the Wedge Clam (Donax trunculus). PeerJ 2017, 18, 3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics, 2nd ed.; Cambridge University Press: Cambridge, UK, 2010; pp. 1–22. [Google Scholar]
- McDermott, J.M.; McDonald, B.A. Gene flow in plant pathosystems. Annu. Rev. Phytopathol. 1993, 31, 353–373. [Google Scholar] [CrossRef]
- Bieber, R.C.; Ruf, T. Population dynamics in wild boar Sus scrofa: Ecology, elasticity of growth rate and implications for the management of pulsed resource consumers. J. Appl. Ecol. 2005, 42, 1203–1213. [Google Scholar] [CrossRef]
- Acevedo, P.; Escudero, M.A.; Muńoz, R.; Gortázar, C. Factors affecting wild boar abundance across an environmental gradient in Spain. Acta. Theriol. 2006, 51, 327–336. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Sites | N | Ar | MAF | Ae | NG | Ap | He | Ho | Fis | PIC | HWE p-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Telšiai | 43 | 9.377 | 0.394 | 5.047 | 17.308 | 7 | 0.742 | 0.601 | 0.201 | 0.718 | 0.000 ** |
| Klaipėda | 32 | 7.954 | 0.431 | 4.194 | 13.000 | 4 | 0.714 | 0.625 | 0.140 | 0.685 | 0.000 ** |
| Tauragė | 56 | 9.392 | 0.397 | 4.630 | 18.615 | 1 | 0.752 | 0.662 | 0.129 | 0.725 | 0.000 ** |
| Marijampolė | 43 | 9.191 | 0.389 | 4.748 | 16.230 | 2 | 0.749 | 0.647 | 0.162 | 0.721 | 0.000 ** |
| Alytus | 28 | 12.638 | 0.293 | 7.188 | 15.615 | 7 | 0.823 | 0.654 | 0.224 | 0.805 | 0.000 ** |
| Kaunas | 56 | 12.347 | 0.283 | 7.286 | 24.154 | 4 | 0.837 | 0.633 | 0.252 | 0.820 | 0.000 ** |
| Vilnius | 53 | 11.985 | 0.321 | 6.370 | 22.385 | 17 | 0.816 | 0.620 | 0.250 | 0.799 | 0.000 ** |
| Šiauliai | 82 | 10.176 | 0.337 | 5.566 | 24.692 | 9 | 0.795 | 0.644 | 0.195 | 0.773 | 0.000 ** |
| Utena | 59 | 11.902 | 0.297 | 6.399 | 24.077 | 9 | 0.826 | 0.568 | 0.320 | 0.809 | 0.000 ** |
| Panevėžys | 39 | 11.711 | 0.331 | 5.757 | 18.000 | 14 | 0.807 | 0.578 | 0.296 | 0.790 | 0.000 ** |
| Mean | - | 10.513 | 0.347 | 5.719 | 19.408 | 6.7 | 0.786 | 0.623 | 0.217 | 0.765 | 0.000 ** |
| SSR Marker | MAF | NA | NG | Ar | He | Ho | PIC | Fis | HWE p-Value | Nm |
|---|---|---|---|---|---|---|---|---|---|---|
| sw24 | 0.310 | 20 | 67 | 11.070 | 0.822 | 0.688 | 0.802 | 0.164 | 0.000 ** | 4.334 |
| s0107 | 0.210 | 29 | 102 | 14.646 | 0.895 | 0.587 | 0.887 | 0.346 | 0.000 ** | 3.836 |
| s0386 | 0.233 | 19 | 60 | 12.795 | 0.892 | 0.684 | 0.883 | 0.234 | 0.000 ** | 4.551 |
| sw72 | 0.388 | 15 | 31 | 7.918 | 0.774 | 0.538 | 0.748 | 0.306 | 0.000 ** | 8.825 |
| tnfb | 0.177 | 33 | 83 | 16.340 | 0.912 | 0.748 | 0.906 | 0.182 | 0.000 ** | 7.152 |
| s0070 | 0.276 | 29 | 59 | 11.825 | 0.858 | 0.646 | 0.845 | 0.249 | 0.000 ** | 4.221 |
| s0026 | 0.333 | 17 | 40 | 10.093 | 0.817 | 0.670 | 0.798 | 0.181 | 0.000 ** | 3.366 |
| s0155 | 0.327 | 20 | 37 | 9.885 | 0.809 | 0.495 | 0.788 | 0.389 | 0.000 ** | 1.705 |
| s0005 | 0.147 | 51 | 179 | 23.046 | 0.944 | 0.798 | 0.942 | 0.155 | 0.000 ** | 6.032 |
| sw2410 | 0.534 | 26 | 46 | 10.243 | 0.683 | 0.405 | 0.664 | 0.408 | 0.000 ** | 2.747 |
| sw830 | 0.418 | 16 | 30 | 7.619 | 0.713 | 0.450 | 0.671 | 0.370 | 0.000 ** | 2.245 |
| sw632 | 0.163 | 20 | 72 | 14.024 | 0.909 | 0.729 | 0.902 | 0.199 | 0.000 ** | 3.912 |
| swr1941 | 0.325 | 19 | 46 | 9.490 | 0.826 | 0.659 | 0.808 | 0.202 | 0.000 ** | 4.761 |
| Mean | 0.295 | 24.153 | 65.53 | 12.230 | 0.835 | 0.623 | 0.819 | 0.255 | 4.437 |
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variation | F-Statistics | Value | p-Value |
|---|---|---|---|---|---|---|---|
| Among sampling sites | 9 | 253.061 | 0.23884 Va | 5.42 | Fst | 0.054 | p < 0.001 |
| Among individuals within sampling sites | 481 | 2358.147 | 0.73236 Vb | 16.61 | Fis | 0.175 | p < 0.001 |
| Within individuals | 491 | 1688.000 | 3.43788 Vc | 77.97 | Fit | 0.220 | p < 0.001 |
| Total | 981 | 4299.208 | 4.40907 | 100.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griciuvienė, L.; Janeliūnas, Ž.; Pilevičienė, S.; Jurgelevičius, V.; Paulauskas, A. Changes in the Genetic Structure of Lithuania’s Wild Boar (Sus scrofa) Population Following the Outbreak of African Swine Fever. Genes 2022, 13, 1561. https://doi.org/10.3390/genes13091561
Griciuvienė L, Janeliūnas Ž, Pilevičienė S, Jurgelevičius V, Paulauskas A. Changes in the Genetic Structure of Lithuania’s Wild Boar (Sus scrofa) Population Following the Outbreak of African Swine Fever. Genes. 2022; 13(9):1561. https://doi.org/10.3390/genes13091561
Chicago/Turabian StyleGriciuvienė, Loreta, Žygimantas Janeliūnas, Simona Pilevičienė, Vaclovas Jurgelevičius, and Algimantas Paulauskas. 2022. "Changes in the Genetic Structure of Lithuania’s Wild Boar (Sus scrofa) Population Following the Outbreak of African Swine Fever" Genes 13, no. 9: 1561. https://doi.org/10.3390/genes13091561