
First Genome of Rock Lizard Darevskia valentini Involved in Formation of Several Parthenogenetic Species

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
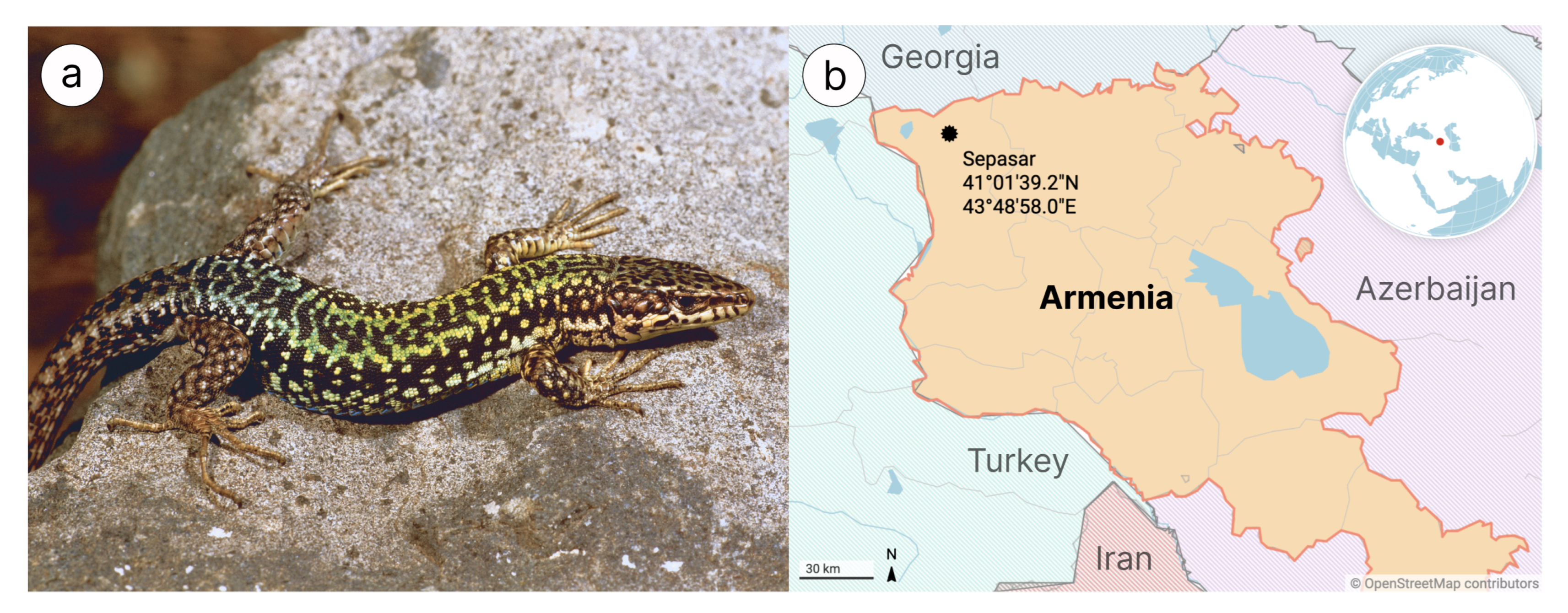
2.1. Samples Collection and Surgical Procedures
2.2. Extraction, Preparation and Sequencing of DNA Libraries
2.3. Transcriptome Libraries Preparation and Sequencing
2.4. Raw Data Preprocessing and Quality Control
2.5. Genome Assembly and Quality Control
2.6. Genome Annotation
2.6.1. Repeats Annotation
2.6.2. Gene Prediction and Functional Annotation
2.6.3. Hox Clusters Annotation
2.7. Phylogenomic Reconstruction
3. Results and Discussion
3.1. Genome Assembly, Validation and Annotation
3.2. Hox Cluster Organisation in D. valentini
3.3. Phylogenomic Reconstruction of Squamate Phylogeny
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Tollis, M.; Hutchins, E.D.; Kusumi, K. Reptile genomes open the frontier for comparative analysis of amniote development and regeneration. Int. J. Dev. Biol. 2014, 58, 863–871. [Google Scholar] [CrossRef]
- Uetz, P.; Freed, P.; Aguilar, R.; Hošek, J. The Reptile Database. 2021. Available online: www.reptile-database.org (accessed on 1 May 2021).
- Arribas, O.J. Phylogeny and Relationships of the Mountain Lizards of Europe and Near East (Archaeolacerta Mertens, 1921, sensu lato) and Their Relationships Among the Eurasian Lacertid Radiation. Russ. J. Herpetol. 1999, 6, 1–22. [Google Scholar]
- Darevsky, I.; Kupriyanova, L.; Uzzell, T. Parthenogenesis in Reptiles. In Biology of Reptilia; John Wiley & Sons: New York, NY, USA, 1985; Volume 15, pp. 413–526. [Google Scholar]
- Neaves, W.B.; Baumann, P. Unisexual reproduction among vertebrates. Trends Genet. 2011, 27, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.W.; Fu, J.; Macculloch, R.D.; Darevsky, I.S.; Kupriyanova, L.A. A fine line between sex and unisexuality: The phylogenetic constraints on parthenogenesis in lacertid lizards. Zool. J. Linn. Soc. 2000, 130, 527–549. [Google Scholar] [CrossRef]
- Fu, J.; Murphy, R.; Darevsky, I. Toward the phylogeny of caucasian rock lizards: Implications from mitochondrial DNA gene sequences (Reptilia: Lacertidae). Zool. J. Linn. Soc. 1997, 120, 463–477. [Google Scholar] [CrossRef]
- Schmidtler, J.; Eiselt, J.; Darevsky, I. Untersuchungen an Felseidechsen (Lacerta saxicola Gruppe) in der östlichen Türkei: 3. Zwei neue parthenogenetische Arten. Salamandra 1994, 30, 55–70. [Google Scholar]
- Ahmad, S.; Singchat, W.; Jehangir, M.; Panthum, T.; Srikulnath, K. Consequence of Paradigm Shift with Repeat Landscapes in Reptiles: Powerful Facilitators of Chromosomal Rearrangements for Diversity and Evolution. Genes 2020, 11, 827. [Google Scholar] [CrossRef]
- Voss, S.R.; Putta, S.; Walker, J.A.; Smith, J.J.; Maki, N.; Tsonis, P.A. Salamander Hox clusters contain repetitive DNA and expanded non-coding regions: A typical Hoxstructure for non-mammalian tetrapod vertebrates? Hum. Genom. 2013, 7, 9. [Google Scholar] [CrossRef]
- Feiner, N. Accumulation of transposable elements in Hox gene clusters during adaptive radiation of Anolis lizards. Proc. R. Soc. B Biol. Sci. 2016, 283, 20161555. [Google Scholar] [CrossRef]
- Di-Poï, N.; Montoya-Burgos, J.I.; Duboule, D. Atypical relaxation of structural constraints in Hox gene clusters of the green anole lizard. Genome Res. 2009, 19, 602–610. [Google Scholar] [CrossRef]
- Kazazian, H.H. Mobile Elements: Drivers of Genome Evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Warren, I.A.; Naville, M.; Chalopin, D.; Levin, P.; Berger, C.S.; Galiana, D.; Volff, J.N. Evolutionary impact of transposable elements on genomic diversity and lineage-specific innovation in vertebrates. Chromosome Res. 2015, 23, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Trizzino, M.; Park, Y.; Holsbach-Beltrame, M.; Aracena, K.; Mika, K.; Caliskan, M.; Perry, G.H.; Lynch, V.J.; Brown, C.D. Transposable elements are the primary source of novelty in primate gene regulation. Genome Res. 2017, 27, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Duboule, D. Guidebook to the Homeobox Genes; Guidebook Series; Oxford University Press: Oxford, UK; New York, NY, USA, 1994. [Google Scholar]
- Holland, P.W.; Garcia-Fernàndez, J.; Williams, N.A.; Sidow, A. Gene duplications and the origins of vertebrate development. Dev. Suppl. 1994, 125–133. [Google Scholar] [CrossRef]
- Lemons, D.; McGinnis, W. Genomic evolution of Hox gene clusters. Science 2006, 313, 1918–1922. [Google Scholar] [CrossRef]
- Liang, D.; Wu, R.; Geng, J.; Wang, C.; Zhang, P. A general scenario of Hox gene inventory variation among major sarcopterygian lineages. BMC Evol. Biol. 2011, 11, 25. [Google Scholar] [CrossRef]
- Feiner, N. Evolutionary lability in Hox cluster structure and gene expression in Anolis lizards. Evol. Lett. 2019, 3, 474–484. [Google Scholar] [CrossRef]
- Lempradl, A.; Ringrose, L. How does noncoding transcription regulate Hox genes? BioEssays 2008, 30, 110–121. [Google Scholar] [CrossRef]
- Petruk, S.; Sedkov, Y.; Brock, H.W.; Mazo, A. A Model for Initiation of Mosaic HOX Gene Expression Patterns by Non-Coding RNAs in Early Embryos. RNA Biol. 2007, 4, 1–6. [Google Scholar] [CrossRef]
- Chen, K.; Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef]
- Tanzer, A.; Amemiya, C.T.; Kim, C.B.; Stadler, P.F. Evolution of microRNAs located withinHox gene clusters. J. Exp. Zool. Part Mol. Dev. Evol. 2005, 304B, 75–85. [Google Scholar] [CrossRef]
- Candiani, S. Focus on miRNAs evolution: A perspective from amphioxus. Briefings Funct. Genom. 2012, 11, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo-Paysaa, F.; Sémon, M.; Cameron, R.A.; Peterson, K.J.; Schubert, M. microRNA complements in deuterostomes: Origin and evolution of microRNAs: miRNA origins and evolution. Evol. Dev. 2011, 13, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.; Lemoine, F.; Soumillon, M.; Liechti, A.; Weier, M.; Guschanski, K.; Hu, H.; Khaitovich, P.; Kaessmann, H. Birth and expression evolution of mammalian microRNA genes. Genome Res. 2013, 23, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Mariño-Ramírez, L.; Jordan, I.K. Origin and Evolution of Human microRNAs from Transposable Elements. Genetics 2007, 176, 1323–1337. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Sun, X.; Liu, H.; Xie, J. MicroRNA Genes Derived from Repetitive Elements and Expanded by Segmental Duplication Events in Mammalian Genomes. PLoS ONE 2011, 6, e17666. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Arakelyan, M.; Danielyan, F.; Corti, C.; Sindaco, R.; Leviton, A.E.; Vasilyan, D. Herpetofauna of Armenia and Nagorno-Karabakh; Number 27 in Contributions to Herpetology; Society for the Study of Amphibians and Reptiles (SSAR): Ithaca, NY, USA, 2011. [Google Scholar]
- Andrews, S. FASTQC. A Quality Control Tool for High Throughput Sequence Data 2010. Available online: https://github.com/s-andrews/FastQC (accessed on 20 September 2021).
- Starostina, E.; Tamazian, G.; Dobrynin, P.; O’Brien, S.; Komissarov, A. Cookiecutter: A tool for kmer-based read filtering and extraction. bioRxiv 2015. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction; Methods in Molecular Biology; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.; Fitzpatrick, D.A. Recent advances in oomycete genomics. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2020; Volume 105, pp. 175–228. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Akam, M. Hox and HOM: Homologous gene clusters in insects and vertebrates. Cell 1989, 57, 347–349. [Google Scholar] [CrossRef]
- Feiner, N.; Wood, N.J. Lizards possess the most complete tetrapod Hox gene repertoire despite pervasive structural changes in Hox clusters. Evol. Dev. 2019, 21, e12300. [Google Scholar] [CrossRef]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef]
- Amemiya, C.T.; Prohaska, S.J.; Hill-Force, A.; Cook, A.; Wasserscheid, J.; Ferrier, D.E.; Pascual-Anaya, J.; Garcia-Fernàndez, J.; Dewar, K.; Stadler, P.F. The amphioxus Hox cluster: Characterization, comparative genomics, and evolution. J. Exp. Zool. Part B Mol. Dev. Evol. 2008, 310B, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Fried, C.; Prohaska, S.J.; Stadler, P.F. Exclusion of repetitive DNA elements from gnathostome Hox clusters. J. Exp. Zool. 2004, 302B, 165–173. [Google Scholar] [CrossRef]
- Kikuta, H.; Fredman, D.; Rinkwitz, S.; Lenhard, B.; Becker, T.S. Retroviral enhancer detection insertions in zebrafish combined with comparative genomics reveal genomic regulatory blocks—A fundamental feature of vertebrate genomes. Genome Biol. 2007, 8, S4. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.K. Structural and sequence diversity of eukaryotic transposable elements. Genes Genet. Syst. 2019, 94, 233–252. [Google Scholar] [CrossRef]
- Sundaram, V.; Wysocka, J. Transposable elements as a potent source of diverse cis -regulatory sequences in mammalian genomes. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190347. [Google Scholar] [CrossRef]
- Petri, R.; Brattås, P.L.; Sharma, Y.; Jönsson, M.E.; Pircs, K.; Bengzon, J.; Jakobsson, J. LINE-2 transposable elements are a source of functional human microRNAs and target sites. PLoS Genet. 2019, 15, e1008036. [Google Scholar] [CrossRef]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-Type Specific Features of Circular RNA Expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Wilusz, J.E. Repetitive elements regulate circular RNA biogenesis. Mob. Genet. Elem. 2015, 5, 39–45. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA Biogenesis Competes with Pre-mRNA Splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Starke, S.; Jost, I.; Rossbach, O.; Schneider, T.; Schreiner, S.; Hung, L.H.; Bindereif, A. Exon Circularization Requires Canonical Splice Signals. Cell Rep. 2015, 10, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Filippenkov, I.B.; Kalinichenko, E.O.; Limborska, S.A.; Dergunova, L.V. Circular RNAs—One of the enigmas of the brain. Neurogenetics 2017, 18, 1–6. [Google Scholar] [CrossRef]
- Lyson, T.R.; Sperling, E.A.; Heimberg, A.M.; Gauthier, J.A.; King, B.L.; Peterson, K.J. MicroRNAs support a turtle + lizard clade. Biol. Lett. 2012, 8, 104–107. [Google Scholar] [CrossRef]
- Garcia-Porta, J.; Irisarri, I.; Kirchner, M.; Rodríguez, A.; Kirchhof, S.; Brown, J.L.; MacLeod, A.; Turner, A.P.; Ahmadzadeh, F.; Albaladejo, G.; et al. Environmental temperatures shape thermal physiology as well as diversification and genome-wide substitution rates in lizards. Nat. Commun. 2019, 10, 4077. [Google Scholar] [CrossRef]
- Pyron, R.A.; Burbrink, F.T.; Wiens, J.J. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol. Biol. 2013, 13, 93. [Google Scholar] [CrossRef]
- Tu, N.; Liang, D.; Zhang, P. Whole-exome sequencing and genome-wide evolutionary analyses identify novel candidate genes associated with infrared perception in pit vipers. Sci. Rep. 2020, 10, 13033. [Google Scholar] [CrossRef]
- Klein, C.G.; Pisani, D.; Field, D.J.; Lakin, R.; Wills, M.A.; Longrich, N.R. Evolution and dispersal of snakes across the Cretaceous-Paleogene mass extinction. Nat. Commun. 2021, 12, 5335. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Value |
|---|---|
| Genome length (bp) | 1,456,729,600 |
| Number of scaffolds | 32,139 |
| Scaffold N50 (bp) | 3,939,878 |
| Scaffold L50 (bp) | 80 |
| %N | 3.73 |
| GC content | 43.96% |
| Category of BUSCOs | eukaryota_odb10 | sauropsida_odb10 |
|---|---|---|
| Number of protein groups in the database | 255 | 7480 |
| Complete | 247 (96.9%) | 6552 (87.6%) |
| Complete and single-copy | 241 (94.5%) | 6313 (84.4%) |
| Complete and duplicated | 6 (2.4%) | 239 (3.2%) |
| Fragmented | 6 (2.4%) | 301 (4.0%) |
| Missing | 2 (0.7%) | 627 (8.4%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochkalova, S.; Korchagin, V.; Vergun, A.; Urin, A.; Zilov, D.; Ryakhovsky, S.; Girnyk, A.; Martirosyan, I.; Zhernakova, D.V.; Arakelyan, M.; et al. First Genome of Rock Lizard Darevskia valentini Involved in Formation of Several Parthenogenetic Species. Genes 2022, 13, 1569. https://doi.org/10.3390/genes13091569
Ochkalova S, Korchagin V, Vergun A, Urin A, Zilov D, Ryakhovsky S, Girnyk A, Martirosyan I, Zhernakova DV, Arakelyan M, et al. First Genome of Rock Lizard Darevskia valentini Involved in Formation of Several Parthenogenetic Species. Genes. 2022; 13(9):1569. https://doi.org/10.3390/genes13091569
Chicago/Turabian StyleOchkalova, Sofia, Vitaly Korchagin, Andrey Vergun, Avel Urin, Danil Zilov, Sergei Ryakhovsky, Anastasiya Girnyk, Irena Martirosyan, Daria V. Zhernakova, Marine Arakelyan, and et al. 2022. "First Genome of Rock Lizard Darevskia valentini Involved in Formation of Several Parthenogenetic Species" Genes 13, no. 9: 1569. https://doi.org/10.3390/genes13091569