
Comparative Transcriptome Analysis of Slow-Twitch and Fast-Twitch Muscles in Dezhou Donkeys


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Selection and Sample Collection
2.2. Histochemical Analysis
2.3. RNA Extraction, Sequencing and Bioinformatics Analysis
2.4. Small RNA Sequencing and miRNA Analysis
2.5. Functional Enrichment Analysis
2.6. Real-time Quantitative PCR
2.7. Protein–Protein Interaction (PPI) Network Analysis
2.8. Statistical Analyses
2.9. Data Availability
3. Results
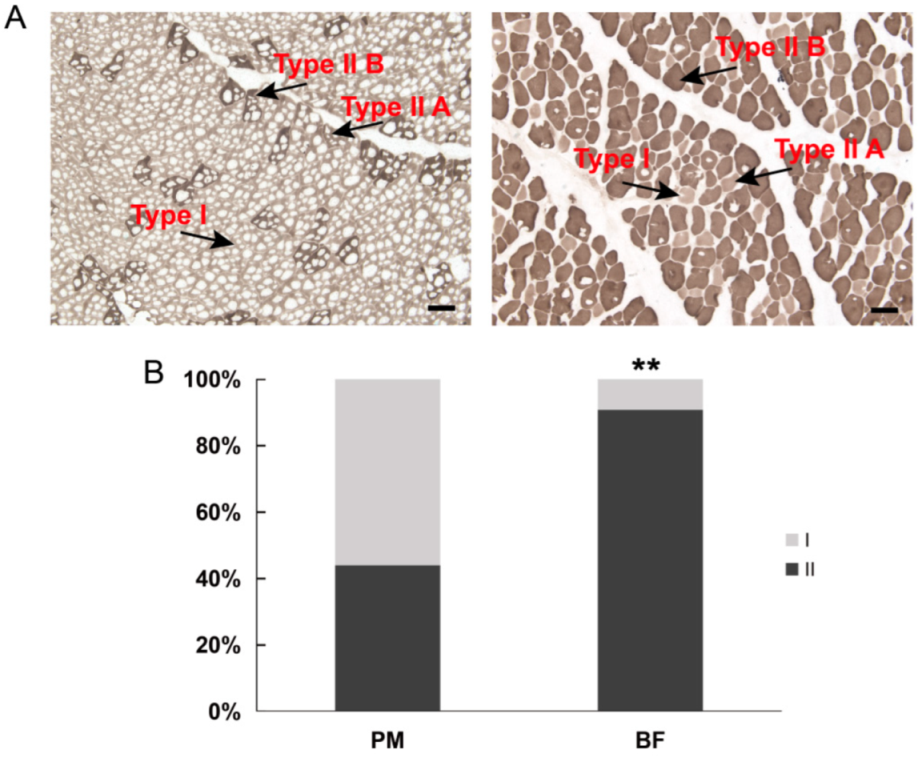
3.1. Muscle Fiber Type Populations
3.2. Overview of RNA and Small-RNA Sequencing
3.3. Differentially Expressed Genes (DEGs) between the Two Muscles
3.4. Differentially Expressed miRNAs (DEmiRs) and Target Gene Prediction
3.5. qRT-PCR Validation
3.6. Combined Analysis of DEGs and DEmiRs
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lorenzo, J.M.; Pateiro, M.; Franco, D. Influence of muscle type on physicochemical and sensory properties of foal meat. Meat Sci. 2013, 94, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-D.; Ryu, Y.-C.; Jeong, J.-Y.; Yang, H.-S.; Joo, S.-T. Relationship between pork quality and characteristics of muscle fibers classified by the distribution of myosin heavy chain isoforms. J. Anim. Sci. 2013, 91, 5525–5534. [Google Scholar] [CrossRef] [PubMed]
- Gaga Oua, M.; Picard, B. Muscle Fiber Properties in Cattle and Their Relationships with Meat Qualities: An Overview. J. Agric. Food Chem. 2020, 68, 6021–6039. [Google Scholar] [CrossRef]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. WIREs Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef]
- Mohammadabadi, M.; Bordbar, F.; Jensen, J.; Du, M.; Guo, W. Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals. Animals 2021, 11, 835. [Google Scholar] [CrossRef]
- Mohammadabadi, M. Expression of calpastatin gene in Raini Cashmere goat using Real Time PCR. Agric. Biotechnol. J. 2019, 11, 219–235. [Google Scholar] [CrossRef]
- Mohammadabadi, M.; Asadollahpour Nanaei, H. Leptin gene expression in Raini Cashmere goat using Real-Time PCR. Agric. Biotechnol. J. 2021, 13, 197–214. [Google Scholar] [CrossRef]
- Chai, W.; Qu, H.; Ma, Q.; Zhu, M.; Li, M.; Zhan, Y.; Liu, Z.; Xu, J.; Yao, H.; Li, Z.; et al. RNA-seq analysis identifies differentially expressed gene in different types of donkey skeletal muscles. Anim. Biotechnol. 2022, 18, 1–10. [Google Scholar] [CrossRef]
- Ouyang, H.; He, X.; Li, G.; Xu, H.; Jia, X.; Nie, Q.; Zhang, X. Deep Sequencing Analysis of miRNA Expression in Breast Muscle of Fast-Growing and Slow-Growing Broilers. Int. J. Mol. Sci. 2015, 16, 16242–16262. [Google Scholar] [CrossRef] [Green Version]
- Mok, G.F.; Lozano-Velasco, E.; Münsterberg, A. microRNAs in skeletal muscle development. Semin. Cell Dev. Biol. 2017, 72, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, M.; Ma, J.; Zhang, J.; Zhou, C.; Wang, T.; Gao, X.; Li, X. Identification of differences in microRNA transcriptomes between porcine oxidative and glycolytic skeletal muscles. BMC Mol. Biol. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Yin, D.; Zhang, L.; Li, B.; Li, R.; Zhang, X.; Zhang, Z.; Liu, H.; Kim, K.; Wu, W. Parsing the microRNA genetics basis regulating skeletal muscle fiber types and meat quality traits in pigs. Anim. Genet. 2021, 52, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, H.; Liu, R.; Jin, L.; Tang, Q.; Wang, X.; Jiang, A.; Hu, Y.; Li, Z.; Zhu, L.; et al. The miRNA Transcriptome Directly Reflects the Physiological and Biochemical Differences between Red, White, and Intermediate Muscle Fiber Types. Int. J. Mol. Sci. 2015, 16, 9635–9653. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, M.; Shan, Y.; Ji, G.; Ju, X.; Tu, Y.; Sheng, Z.; Xie, J.; Zou, J.; Shu, J. miRNA-mRNA network regulation in the skeletal muscle fiber phenotype of chickens revealed by integrated analysis of miRNAome and transcriptome. Sci. Rep. 2020, 10, 10619. [Google Scholar] [CrossRef]
- Wang, X.Y.; Chen, X.L.; Huang, Z.Q.; Chen, D.W.; Yu, B.; He, J.; Luo, J.Q.; Luo, Y.H.; Chen, H.; Zheng, P.; et al. MicroRNA-499-5p regulates porcine myofiber specification by controlling Sox6 expression. Animal 2017, 11, 2268–2274. [Google Scholar] [CrossRef]
- Shen, L.; Chen, L.; Zhang, S.; Zhang, Y.; Wang, J.; Zhu, L. MicroRNA-23a reduces slow myosin heavy chain isoforms composition through myocyte enhancer factor 2C (MEF2C) and potentially influences meat quality. Meat Sci. 2016, 116, 201–206. [Google Scholar] [CrossRef]
- Bao, T.; Han, H.; Li, B.; Zhao, Y.; Bou, G.; Zhang, X.; Du, M.; Zhao, R.; Mongke, T.; Laxima; et al. The distinct transcriptomes of fast-twitch and slow-twitch muscles in Mongolian horses. Comp. Biochem. Phys. D 2020, 33, 100649. [Google Scholar] [CrossRef]
- Zhao, C.; Teng, J.; Zhang, X.; Wang, D.; Zhang, X.; Li, S.; Li, H.; Jiang, X.; Ning, C.; Zhang, Q. Optimizing Genomic Selection in Dezhou Donkey Using Low Coverage Whole Genome Sequencing. Res. Sq. 2021, preprint. [Google Scholar] [CrossRef]
- Li, M.; Zhang, D.; Chai, W.; Zhu, M.; Wang, Y.; Liu, Y.; Wei, Q.; Fan, D.; Lv, M.; Jiang, X.; et al. Chemical and physical properties of meat from Dezhou black donkey. Food Sci. Technol. Res. 2022, 28, 87–94. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Huang, B.; Zhu, M.; Wang, C. The Fibrolytic Enzyme Profiles and the Composition of Fungal Communities in Donkey Cecum-Colon Ecosystem. Animals 2022, 12, 412. [Google Scholar] [CrossRef] [PubMed]
- Brooke, M.H.; Kaiser, K.K. Muscle Fiber Types: How Many and What Kind? Arch. Neurol. 1970, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, A.B.; Smith, G.R.; Raue, U.; Begue, G.; Minchev, K.; Ruf-Zamojski, F.; Nair, V.D.; Wang, X.; Zhou, L.; Zaslavsky, E.; et al. Single-cell transcriptional profiles in human skeletal muscle. Sci. Rep. 2020, 10, 229. [Google Scholar] [CrossRef]
- Ke, Y.; Mitacek, R.M.; Abraham, A.; Mafi, G.G.; VanOverbeke, D.L.; DeSilva, U.; Ramanathan, R. Effects of muscle-specific oxidative stress on cytochrome c release and oxidation–reduction potential properties. J. Agric. Food Chem. 2017, 65, 7749–7755. [Google Scholar] [CrossRef]
- Shen, L.; Gan, M.; Chen, L.; Zhao, Y.; Niu, L.; Tang, G.; Jiang, Y.; Zhang, T.; Zhang, S.; Zhu, L. miR-152 targets pyruvate kinase to regulate the glycolytic activity of pig skeletal muscles and affects pork quality. Meat Sci. 2022, 185, 108707. [Google Scholar] [CrossRef]
- Lang, Y.; Zhang, S.; Xie, P.; Yang, X.; Sun, B.; Yang, H. Muscle fiber characteristics and postmortem quality of longissimus thoracis, psoas major and semitendinosus from Chinese Simmental bulls. Food Sci. Nutr. 2020, 8, 6083–6094. [Google Scholar] [CrossRef]
- Hwang, Y.-H.; Joo, S.-H.; Bakhsh, A.; Ismail, I.; Joo, S.-T. Muscle fiber characteristics and fatty acid compositions of the four major muscles in Korean native black goat. Korean J. Food Sci. Anim. Resour. 2017, 37, 948–954. [Google Scholar] [CrossRef]
- Li, R.; Li, B.; Jiang, A.; Cao, Y.; Hou, L.; Zhang, Z.; Zhang, X.; Liu, H.; Kim, K.-H.; Wu, W. Exploring the lncRNAs Related to Skeletal Muscle Fiber Types and Meat Quality Traits in Pigs. Genes 2020, 11, 883. [Google Scholar] [CrossRef]
- Fang, M.; Cui, R.; Kang, X.; Liu, Y.; Li, Z.; Liu, X.; Chan, S.; Wang, Y. Integrated analysis of the whole-transcriptome of sheep skeletal muscle reveals the ceRNA regulation network related to muscle fiber formation in sheep. Res. Sq. 2022, preprint. [Google Scholar] [CrossRef]
- Hou, X.; Liu, Q.; Meng, Q.; Wang, L.; Yan, H.; Zhang, L.; Wang, L. TMT-based quantitative proteomic analysis of porcine muscle associated with postmortem meat quality. Food Chem. 2020, 328, 127133. [Google Scholar] [CrossRef]
- Dos Santos, M.; Backer, S.; Auradé, F.; Wong, M.M.-K.; Wurmser, M.; Pierre, R.; Langa, F.; Do Cruzeiro, M.; Schmitt, A.; Concordet, J.-P.; et al. A fast Myosin super enhancer dictates muscle fiber phenotype through competitive interactions with Myosin genes. Nat. Commun. 2022, 13, 1039. [Google Scholar] [CrossRef]
- Yang, W. Structural basis of PKM2 regulation. Protein Cell 2015, 6, 238–240. [Google Scholar] [CrossRef]
- Kim, G.-D.; Jeong, J.-Y.; Yang, H.-S.; Hur, S.J. Differential abundance of proteome associated with intramuscular variation of meat quality in porcine longissimus thoracis et lumborum muscle. Meat Sci. 2019, 149, 85–95. [Google Scholar] [CrossRef]
- Wu, P.; Chen, L.; Cheng, J.; Pan, Y.; Zhu, X.; Bao, L.; Chu, W.; Zhang, J. The miRNA expression profile directly reflects the energy metabolic differences between slow and fast muscle with nutritional regulation of the Chinese perch (Siniperca chuatsi). Comp. Biochem. Phys. A 2021, 259, 111003. [Google Scholar] [CrossRef]
- Hamill, R.M.; McBryan, J.; McGee, C.; Mullen, A.M.; Sweeney, T.; Talbot, A.; Cairns, M.T.; Davey, G.C. Functional analysis of muscle gene expression profiles associated with tenderness and intramuscular fat content in pork. Meat Sci. 2012, 92, 440–450. [Google Scholar] [CrossRef]
- Liu, J.; Liang, X.; Zhou, D.; Lai, L.; Xiao, L.; Liu, L.; Fu, T.; Kong, Y.; Zhou, Q.; Vega, R.B. Coupling of mitochondrial function and skeletal muscle fiber type by a miR-499/Fnip1/AMPK circuit. EMBO Mol. Med. 2016, 8, 1212–1228. [Google Scholar] [CrossRef]
- Zhang, P.; Du, J.; Guo, X.; Wu, S.; He, J.; Li, X.; Shen, L.; Chen, L.; Li, B.; Zhang, J.; et al. LncMyoD Promotes Skeletal Myogenesis and Regulates Skeletal Muscle Fiber-Type Composition by Sponging miR-370-3p. Genes 2021, 12, 589. [Google Scholar] [CrossRef]
- Zhao, Q.; Kang, Y.; Wang, H.Y.; Guan, W.J.; Li, X.C.; Jiang, L.; He, X.H.; Pu, Y.B.; Han, J.L.; Ma, Y.H.; et al. Expression profiling and functional characterization of miR-192 throughout sheep skeletal muscle development. Sci. Rep. 2016, 6, 30281. [Google Scholar] [CrossRef] [PubMed]
- Shang, Q.; Shen, G.; Chen, G.; Zhang, Z.; Yu, X.; Zhao, W.; Zhang, P.; Chen, H.; Tang, K.; Yu, F.; et al. The emerging role of miR-128 in musculoskeletal diseases. J. Cell. Physiol. 2021, 236, 4231–4243. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Pan, W.; Fedulov, A.V.; Jester, W.; Jones, M.R.; Weiss, S.T.; Panettieri, R.A., Jr.; Tantisira, K.; Lu, Q. MicroRNA-10a controls airway smooth muscle cell proliferation via direct targeting of the PI3 kinase pathway. FASEB J. 2014, 28, 2347–2357. [Google Scholar] [CrossRef]
- Jammal, R.; Krowiorz, K.; Haetscher, N.; Emmrich, S.; Rouhi, A.; Heuser, M.; Bothur, S.; Bullinger, L.; Döhner, K.; Lai, C.; et al. The miRNA-193 Family Is a Potent Tumor-Suppressor and a Biomarker for Poor Prognosis in Acute Myeloid Leukemia. Blood 2016, 128, 1534. [Google Scholar] [CrossRef]
- Xu, J.H.; Zhao, J.X.; Jiang, M.Y.; Yang, L.P.; Sun, M.L.; Wang, H.W. MiR-193 promotes cell proliferation and invasion by ING5/PI3K/AKT pathway of triple-negative breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3122–3129. [Google Scholar] [CrossRef]
- Han, F.; Zhou, L.; Zhao, L.; Wang, L.; Liu, L.; Li, H.; Qiu, J.; He, J.; Liu, N. Identification of miRNA in Sheep Intramuscular Fat and the Role of miR-193a-5p in Proliferation and Differentiation of 3T3-L1. Front. Genet. 2021, 12, 633295. [Google Scholar] [CrossRef]
- Ju, X.; Liu, Y.; Shan, Y.; Ji, G.; Zhang, M.; Tu, Y.; Zou, J.; Chen, X.; Geng, Z.; Shu, J. Analysis of potential regulatory LncRNAs and CircRNAs in the oxidative myofiber and glycolytic myofiber of chickens. Sci. Rep. 2021, 11, 20861. [Google Scholar] [CrossRef]
- Hettige, P.; Tahir, U.; Nishikawa, K.C.; Gage, M.J. Comparative analysis of the transcriptomes of EDL, psoas, and soleus muscles from mice. BMC Genom. 2020, 21, 808. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Ma, Q.; Shi, X.; Yuan, W.; Liu, G.; Wang, C. Comparative Transcriptome Analysis of Slow-Twitch and Fast-Twitch Muscles in Dezhou Donkeys. Genes 2022, 13, 1610. https://doi.org/10.3390/genes13091610
Li Y, Ma Q, Shi X, Yuan W, Liu G, Wang C. Comparative Transcriptome Analysis of Slow-Twitch and Fast-Twitch Muscles in Dezhou Donkeys. Genes. 2022; 13(9):1610. https://doi.org/10.3390/genes13091610
Chicago/Turabian StyleLi, Yan, Qingshan Ma, Xiaoyuan Shi, Wenmin Yuan, Guiqin Liu, and Changfa Wang. 2022. "Comparative Transcriptome Analysis of Slow-Twitch and Fast-Twitch Muscles in Dezhou Donkeys" Genes 13, no. 9: 1610. https://doi.org/10.3390/genes13091610
APA StyleLi, Y., Ma, Q., Shi, X., Yuan, W., Liu, G., & Wang, C. (2022). Comparative Transcriptome Analysis of Slow-Twitch and Fast-Twitch Muscles in Dezhou Donkeys. Genes, 13(9), 1610. https://doi.org/10.3390/genes13091610