Abstract
Orchardgrass (Dactylis glomerata L.) is a species in the Gramineae family that is highly important economically and valued for its role in ecology. However, the phylogeny and taxonomy of D. glomerata are still controversial based on current morphological and molecular evidence. The study of chloroplast (cp) genomes has developed into a powerful tool to develop molecular markers for related species and reveal the relationships between plant evolution and phylogenetics. In this study, we conducted comparative genomic analyses and phylogenetic inferences on 14 cp genomes of D. glomerata originating from the Mediterranean and Eurasia. The genome size ranged from 134,375 bp to 134,993 bp and exhibited synteny of gene organization and order. A total of 129–131 genes were identified, including 85–87 protein coding genes, 38 tRNA genes and 8 rRNA genes. The cp sequences were highly conserved, and key sequence variations were detected at the junctions of inverted repeats (IRs)/small single–copy (SSC) regions. Moreover, nine highly variable regions were identified among the subspecies based on a sequence divergence analysis. A total of 285 RNA editing sites were detected that were relevant to 52 genes, where rpoB exhibited the most abundant RNA editing sites. The phylogenetic analysis revealed that all Dactylis subspecies clustered into a monophyletic group and most branches provided a high support bootstrap. The main divergence time of D. glomerata was dated to the Miocene era, and this could have been due to changes in the climate. These findings will provide useful insights for further studies on phylogeny, the identification of subspecies and the development of hypotheses for the evolutionary history of the genus Dactylis and of the Gramineae family.
1. Introduction
Orchardgrass (Dactylis glomerata L.) is a member of the Gramineous family [1]. The genus contains one species and numerous subspecies that have different ploidy levels [2]. Orchardgrass is native to Eurasia and northern Africa, although it has been introduced to nearly every continent and utilized as excellent cool–season forage for the livestock industry that requires forage [3]. In China, orchardgrass has already become an elite forage crop for mixed pasture construction and rocky desertification improvement in southwestern regions. It is highly valued economically and has a strong potential for utilization, in addition to its substantial ecological importance [4]. Based on ploidy, there are primarily three categories of the genus Dactylis: diploid (2n = 2x = 14), tetraploid (2n = 2x = 28) and hexaploid (2n = 2x = 42) [5]. Tetraploid populations are widely used in forage production, while hexaploid populations are rarely reported [6]. For a long time, taxonomists focused on the evolutionary relationship between orchardgrass diploids and tetraploids. Nevertheless, the absence of taxonomically diagnostic characters, and the high morphological similarity among the species, has made it difficult to classify the genus [7]. Although previous research has attempted to classify the Dactylis genus at the cytological and genetic levels, a unified standard for the taxonomy of the subspecies is still lacking. Dactylis classification has been repeatedly revised based on herbaria and field studies [8], cytology and genetics [9]. Molecular genetic analysis of partial DNA sequencing recently [2,5] changed clustering from three (a diploid and two tetraploid groups) to two (comprising, respectively, 17 diploids and six tetraploids). In one sense, although these studies have greatly improved the understanding of the taxonomy and phylogeny of the Dactylis genus, controversy about the phylogenetic status and interspecific phylogenetic relationship of the Dactylis genus remains.
The chloroplast (cp) is a key organelle with substantial functions in photosynthesis, carbon fixation, translation, and transcription [10,11]. Typically, a cp has a highly conserved genome sequence that contains 100–130 genes with a range of sizes from 120–170 kb in most land plants [12,13]. A cp genome usually has a covalently closed circular molecular structure that contains two inverted repeats (IRs) separated by a large single–copy (LSC) region and a small single–copy (SSC) region [14]. Compared with nuclear genomes, cp genomes are characterized by a highly conserved genome structure, a moderate substitution rate and uniparental inheritance, which have been widely used for plant phylogeny, and to identify species, estimate divergence and generate genetic markers [15,16]. In recent years, rapid advances in next–generation sequencing (NGS) technologies have enabled many studies with high–quality genomes with raw reads—such genome sequencing generates genome sequences much more rapidly and economically than using traditional Sanger sequencing [17,18]. Therefore, we have witnessed a greatly increased number of complete plant cp genomes in recent years. To date, more than 6500 complete cp genome sequences have become available in the National Center for Biotechnology Information (NCBI) since the first cp genome, that of tobacco (Nicotiana tabacum), was sequenced in 1986 [19].
However, there have been few studies on the comparative analysis of cp genomes in the Dactylis genus to date. In this study, we sequenced and assembled complete cp genomes of 14 subspecies of D. glomerata using next–generation sequencing. Subsequently, we analyzed the structural characteristics of the genome and identified the variant regions and RNA editing sites in the cp genomes. In addition, the divergence date was estimated, and phylogenetic relationships were reconstructed to assess the taxonomic positions of Dactylis species. This study aims to provide a reference for the taxonomy, phylogeny, and population genetics of D. glomerata. The research should facilitate the exploration and utilization of forage resources.
2. Materials and Methods
2.1. Plant Material and DNA Extraction
Fresh leaves of 14 subspecies of D. glomerata were collected from the Sichuan Agricultural University experimental greenhouse (30°42′ N, 103°51′ E; Chengdu, China). The lighting was set to 14 h/10 h (day/night) with a temperature of 22 °C/15 °C (day/night) (Table S1). The total genomic DNA was extracted from 100 mg of fresh leaves using a Plant Genomic DNA Kit (Tiangen, Beijing, China).
2.2. Chloroplast Genome Sequencing, Assembly and Annotation
The fragmented DNA was prepared for libraries using a VAHTS Universal DNA Library Prep Kit for Illumina V3 (Jisi Huiyuan Biotechnologies Co., Ltd., Nanjing, China); the sequencing read length was PE150. The libraries that passed quality inspection were sequenced on an Illumina Nova Seq 6000 Platform (San Diego, CA, USA). The raw reads were filtered with a threshold of average quality <Q5 and a number of N > 5 using the fastp tool (version 0.20.0, https://github.com/OpenGene/fastp, accessed on 1 April 2022) [20]. The cp genomes of 14 Dactylis subspecies were assembled using the SPAdes pipeline (v3.10.1) [21]. To annotate the cp genome coding sequences (CDS), rRNA and tRNA were acquired using prodigal v2.6.3 (http://www.github.com/hyattpd/Prodigal, accessed on 1 April 2022) [22], hmmer v3.1b2 (http://www.hummer.org/, accessed on 1 April 2022) [23] and ARAGORN v1.2.38 (http://130.235.244.92/ARAGORN accessed on 1 April 2022) [24]. In addition, cp genomic data were extracted from NCBI using BLAST v2.6 (http://blast.ncbi.nlm.nih.gov/Blast.cqi, accessed on 1 April 2022) to align the assembled sequences [25]. The annotation results were then manually checked, and incorrect annotations were removed, along with redundant annotations. The boundaries of multiple exons were determined. Finally, circle maps of all the cp genomes were drawn using the program OGDRAW v1.1 [26].
2.3. Genome Comparison
To compare the structure of the cp genomes of all the D. glomerata subspecies, the borders between the IR and SC regions were analyzed. The program mVISTA was used to compare the cp genomes of 14 Dactylis subspecies, with the annotation of D. aschersoniana used as the reference [27]. Mauve (v2.3.1) software was used to analyze the homology and collinearity to align the genome [28].
2.4. Repeat Element Analysis and RNA Editing Identification
MISA (Microsatellite Identification Tool, v1.0) software was used to analyze the cpSSR using parameters 1–8 (single base repeat 8 times or more) [29]. The software Vmatch combined with a perl script was used to identify repeat sequences [30]. The parameters were as follows: minimum length = 30 bp, Hamming distance = 3, and the four identification forms were forward, palindromic, reverse, and complement. The editing sites of genes among the 14 subspecies of D. glomerata were predicted using PREP–CP with a cutoff value of 0.8 (http://prep.unl.edu/cgi-bin/cp-input.pl, accessed on 1 April 2022) [31].
2.5. Phylogenetic Analysis and the Estimation of Divergence Time
The cp genome sequences were used to construct a phylogenetic tree. The interspecies sequences were aligned by MAFFT software (v7.427–auto mode), and trimAl (v1.4. rev 15) was used to trim the well–aligned data [32]. Using RAxML software (v8.2.10) [33], a GTAGAMMA model with bootstrap analysis executed with 1000 replicates was used to construct the maximum likelihood evolutionary tree. Based on the phylogenetic tree obtained, mcmctree software in the paml (v4.9) package was used to construct the phylogenetic tree of divergence time, and the fossil time of species was searched from the timetree website (http://www.timetree.org/, accessed on 1 April 2022) [34]. Time calibration was based on fossil records of Oryza sativa, with a confidence range of 42–52 Mya. Trapa and Corynocarpus served as outgroups.
3. Results
3.1. Features of the Cp Genomes of the 14 Subspecies of D. glomerata
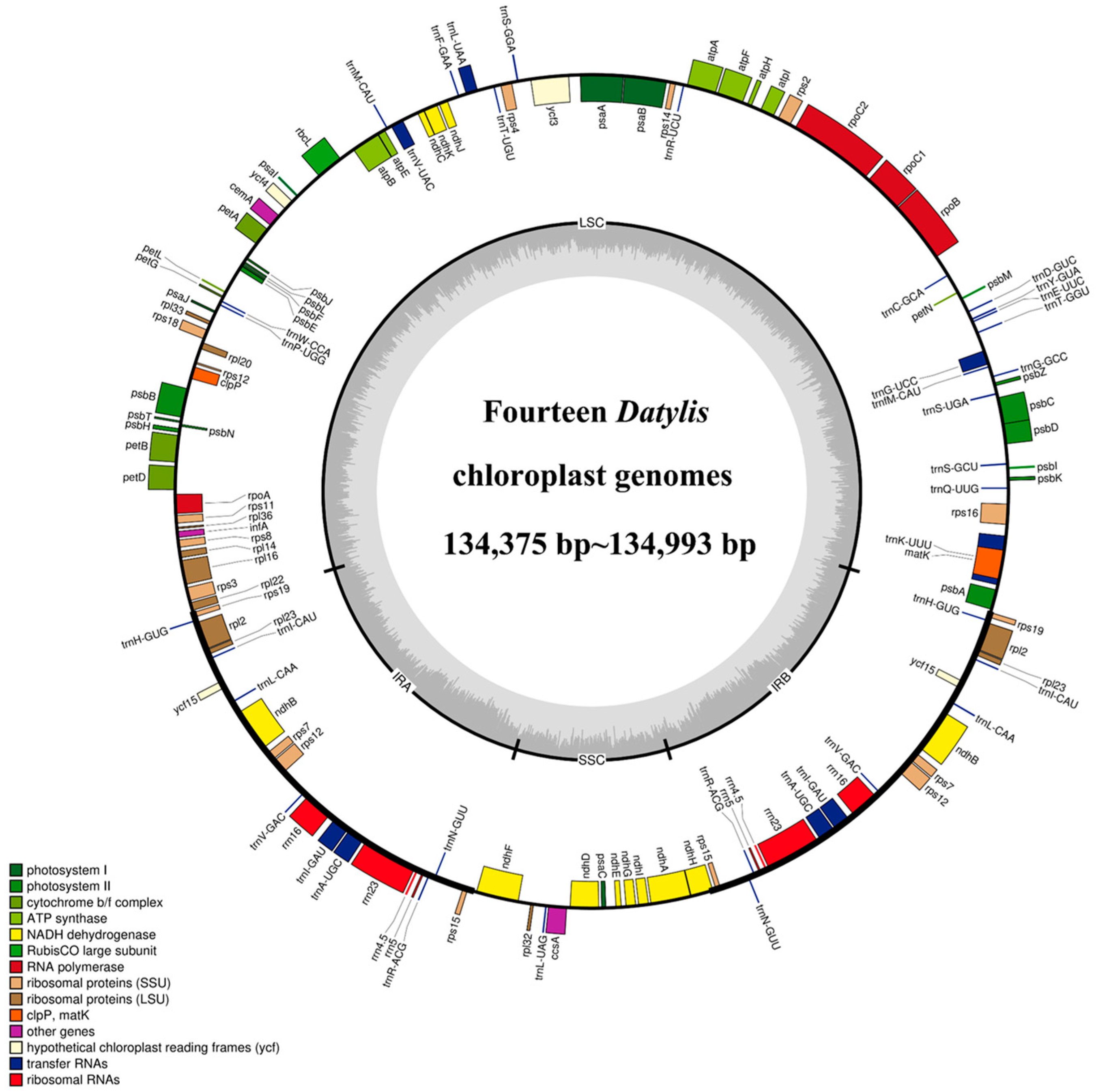
All the cp genomes of D. glomerata subspecies contained a typical quadripartite structure, comprising two IR (IRA and IRB) regions separated by a long single copy (LSC) and a small single copy (SSC) region. The length of the cp genomes of 14 subspecies of D. glomerata ranged from 134,375 bp (D. glomerata subsp. lobata) to 134,993 bp (D. glomerata subsp. lusitanica) (Figure 1). The lengths of LSC, SSC and IRs ranged from 79,753 to 79,773 bp, 12,246 to 12,276 bp and from 21,236 to 21,479 bp, respectively. The size of the LSC, SSC and IRs, as well as those of the whole cp genomes, were shorter than those of others in the two cp genomes of the subspecies D. glomerata subsp. lobata and D. glomerata subsp. hispanica.1 (Table S2). The total GC content was 38–38.44%, while the average GC contents of the LSC, SSC and IR regions were 36.32%, 32.86% and 43.94%, respectively (Table S2). Moreover, a total of 129 genes were identified in the D. glomerata subspecies, excluding 131 genes identified in D. glomerata subsp. hispanica.1. The number of genes encoding the rRNA and tRNA was highly conserved, with eight genes encoding rRNA, and 38 genes encoding tRNA. The cp genomes of D. glomerata contained 83 protein–coding genes, while 85 protein–coding genes were identified in the cp genomes of D. glomerata subsp. hispanica.1 (Table S2). Eight tRNA genes (trnH–GUG, trnI–CAU, trnL–CAA, trnV–GAC, trnI–GAU, trnA–UGC, trnR–ACG and trnN–GUU), four rRNA (rrn4.5, rrn5, rrn16 and rrn23), and seven protein–coding genes (rps19, rpl2, rpl23, ndhB, rps7, rps12 and rps15) were duplicated in the IRs, excluding D glomerata subsp. hispanica.1 (Figure 1). A putative gene of unknown function, ycf15, was present only in IRs in D. glomerata subsp. hispanica.1 among 14 subspecies of D. glomerata (Table S3). Interestingly, we found that a unique gene, infA, was distributed in all Dactylis subspecies. A total of 21 intron–containing genes were found in the cp genomes of all the subspecies, including 13 protein–coding genes and eight tRNA genes (Table S4). Twelve protein–coding genes and eight tRNA genes contained one intron, and two genes (ycf3 and rps12) contained two introns. Slight differences were found in the sizes of introns between different types of D. glomerata subspecies, as shown in Table S4. In all the cp genomes of D. glomerata subspecies, the trnK–UUU intron, including matK, contained the longest introns (2497–2523 bp).
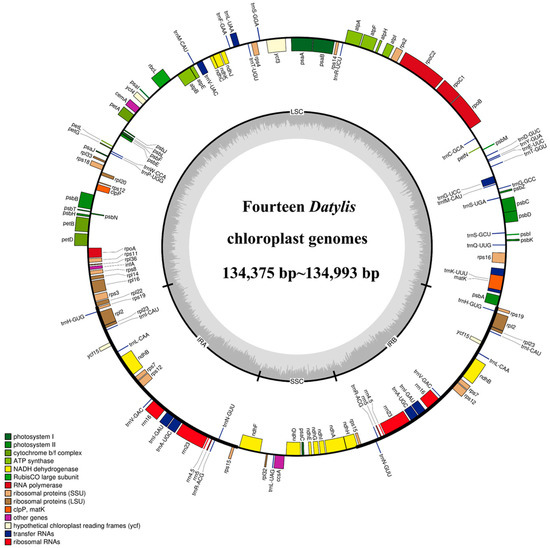
Figure 1.
Representative cp genome of Dactylis subspecies. Genes drawn inside and outside of the circle are transcribed in clockwise and counter–clockwise directions, respectively. The colored bar indicates chloroplast gene groups. The dark gray bar graphs inner circle shows the GC content, and the light gray bar graphs show the AT content.
3.2. Comparative Genome Analysis
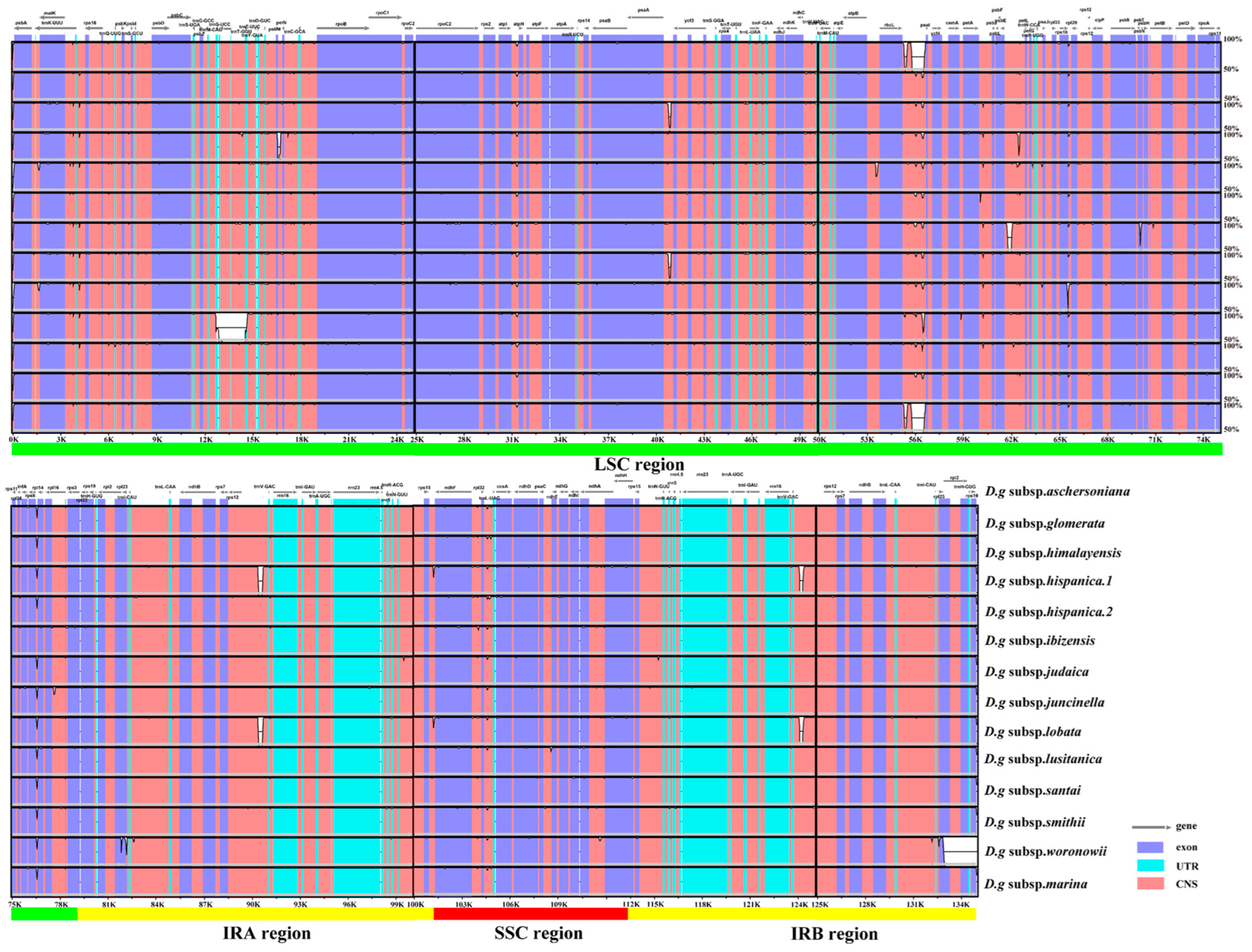
Fourteen cp genomes of the D. glomerata subspecies had highly similar sequences (Figure 2). The IR regions were more conserved than the SC regions. Among these cp genomes, the sequences in coding regions were almost identical or nearly identical. On the other hand, sequences in the noncoding regions were highly variable relative to the sequences in coding regions. The highly divergent regions were found in the intergenic spacers and introns, including regions of rps19–psbA, psbM–petN, trnG–UCC–trnT–GGU, psaA–ycf3, rbcL–psal, psbE–petL, rps12–trnV–GAC, trnV–GAC–rps12 and rpl2–rpl23. These highly divergent regions have the potential to be used for discrimination or phylogeny investigations of the 14 subspecies. Furthermore, we checked the possible rearrangement events, which indicated that the genome structures and gene sequences were basically identical, and no gene rearrangement had occurred (Figure S1).
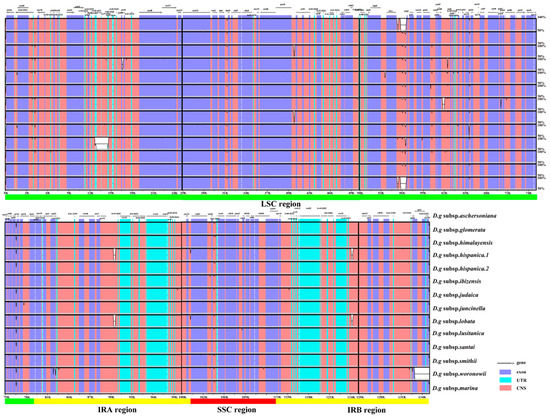
Figure 2.
Sequence identity plot for cp genomes of Dactylis subspecies with D.g subsp. aschersoniana, as reference visualized by mVISTA. The gray arrows indicate the orientations of genes, the red blocks represent the intergenic region, the purple blocks represent exons, the blue blocks represent untranslated regions (UTRs), and the Y–axis represents the percent identity within 50–100%.
3.3. IR Contraction and Expansion
The contraction and expansion of the chloroplast IR regions appear to have a substantial role in the process of plant evolution, which is regarded as the main reason for the variation in genome size and gene quantity in different plants. Among the 14 subspecies of D. glomerata, D. glomerata subsp. lusitanica showed the longest cp genome (134,993 bp), with an IR of 21,472 bp, while the shortest cp genome sizes (134,375–134,376 bp) and IRs (21,236 bp) were detected in two subspecies, D. glomerata subsp. lobata and D. glomerata subsp. hispanica.1 (Figure 3). We compared the IR/SC junctions of the 14 subspecies of D. glomerata and found that the IR/SSC junctions varied slightly. The location of ndhF was 109–110 downstream of the IRb–SSC junction.
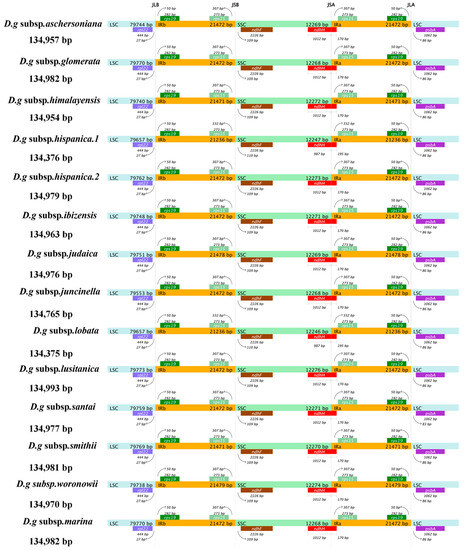
Figure 3.
Comparison of the boundaries of LSC, SSC, and IR regions within cp genomes of 14 D. glomerata subspecies. Genes are denoted by bars, the gaps between the genes and the boundaries are indicated by the base lengths (bp). Extensions of the genes are indicated above the bars.
The SSC–IRa junction was located within the coding region of ndhH, and the partial sequence of ndhH was 1012 bp within the SSC region, while only 987 bp of ndhH was found within the SSC region in the subspecies D. glomerata subsp. lobata and D. glomerata subsp. hispanica.1. The sequence length of ndhH, which was located in IRa, was 170 bp, except in D. glomerata subsp. lobata and D. glomerata subsp. hispanica.1. Correspondingly, rps15 was located 332 bp upstream of the IRa/SSC junction in D. glomerata subsp. lobata and D. glomerata subsp. hispanica.1, which had a 25 bp difference from the other 12 subspecies.
3.4. Characterization of SSRs and Repeat Sequences
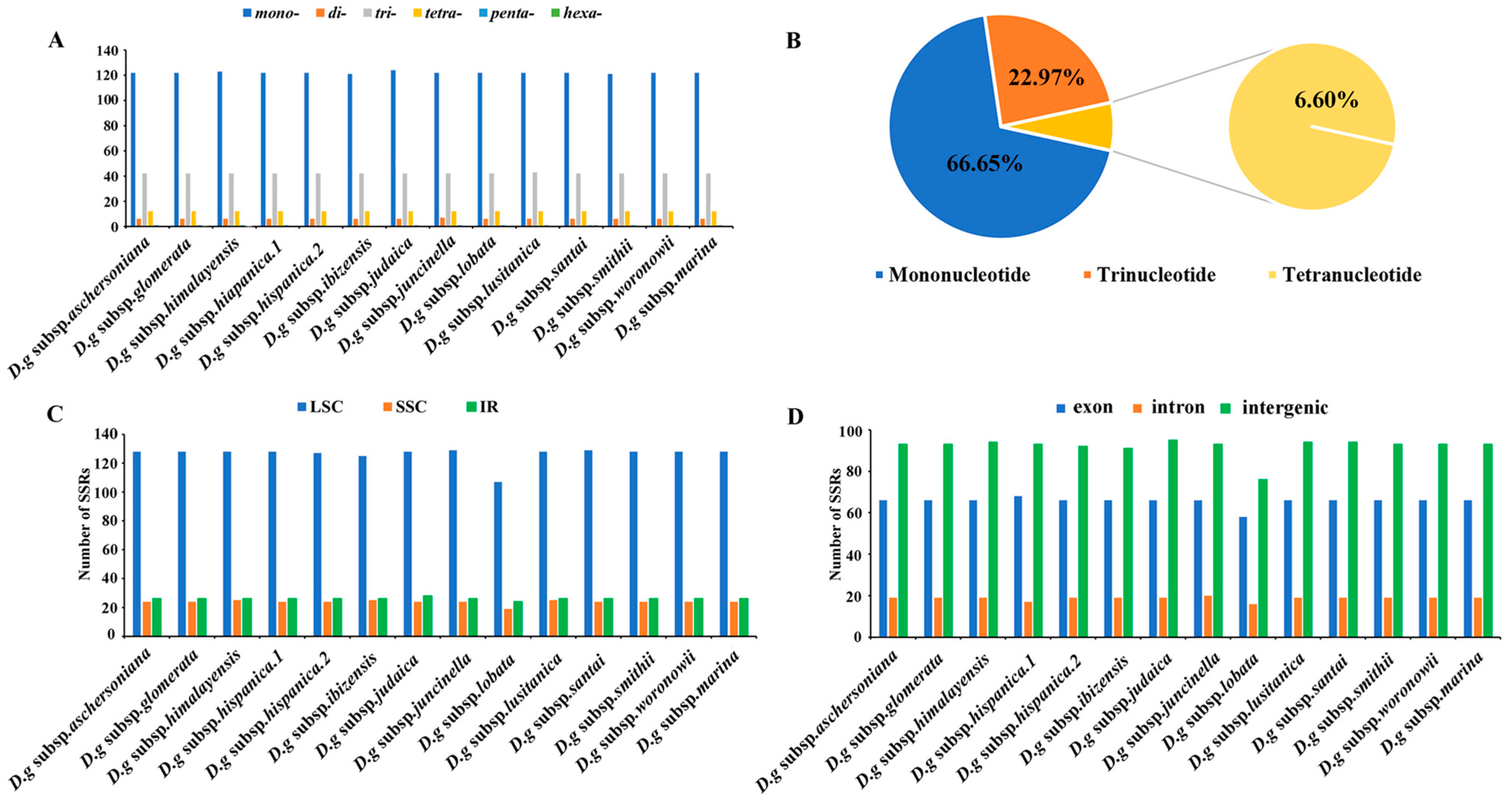
Six categories of SSRs were detected in the cp genomes of 14 subspecies of D. glomerata (Figure 4). The number of cpSSRs varied in all the subspecies and ranged from 181 (D. glomerata subsp. ibizensis) to 185 (D. glomerata subsp. judaica). Most of the SSRs were mononucleotide SSRs, which comprised 66.65% of the total SSRs, followed by trinucleotide (22.97%) and tetranucleotide repeats (6.60%) (Figure 4B). Moreover, the number of tetranucleotide repeats was higher than the number of pentanucleotide and dinucleotide repeats. Pentanucleotide repeats were found in the cp genomes of 12 subspecies of D. glomerata except for D. glomerata subsp. hispanica.2 and D. glomerata subsp. ibizensis. Notably, the hexanucleotide repeats were exceedingly rare across the cp genomes of these subspecies, and only one was identified in the cp genome of D. glomerata subsp. santai (Figure 4A). Most of the SSRs were distributed in the LSC regions, followed by the IR and SSC regions (Figure 4C). In contrast to the exon and intron regions, most of the SSRs were detected in the intergenic regions (Figure 4D). Moreover, most of the SSRs were comprised of the repeat type A/T, rather than the repeat type G/C (Table S5).
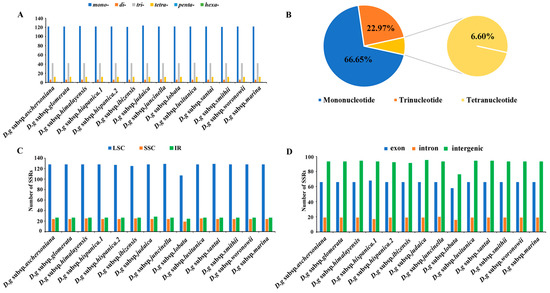
Figure 4.
Analysis of simple sequence repeats (SSRs) in the cp genomes of 14 D. glomerata subspecies. (A) Number of different SSR types detected in cp genomes of D. glomerata subspecies; (B) The proportion of mononucleotide, trinucleotide and tetranucleotide SSRs; (C) Number of SSRs in LSC, SSC and IR regions; (D) Number of SSRs in exon, intron, and intergenic regions.
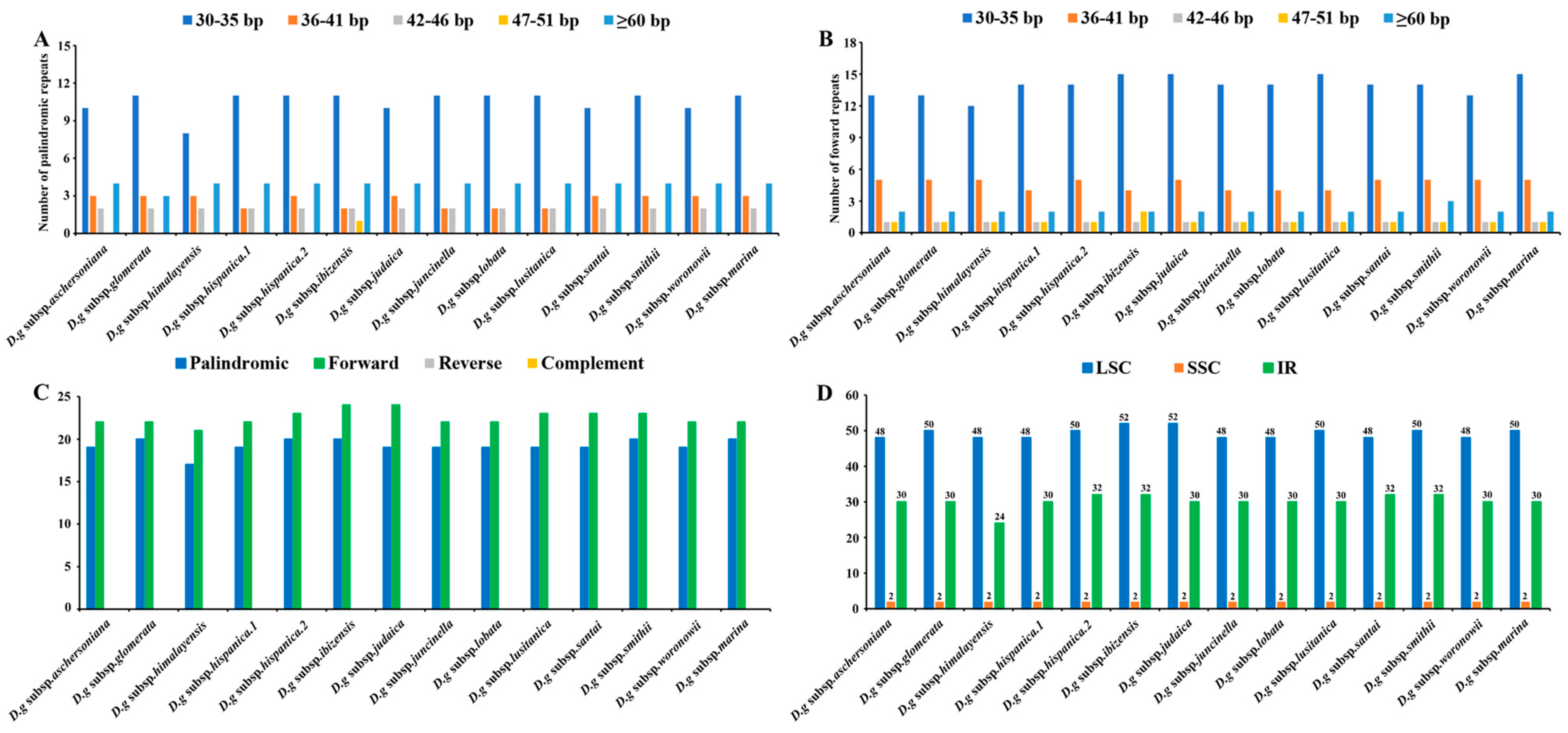
Interspersed repeated sequences are a type of repeat that differ from tandem repeats, which are distributed in a decentralized manner in the genome. Typically, interspersed repeats are classified into four types: forward, palindromic, reverse and complement repeats. In this study, we merely found palindromic repeats and forward repeats, which were similar to the number of interspersed repeats in all the subspecies (Figure 5A,B). The number of interspersed repeats varied from 38 to 44 in all the subspecies. D. glomerata subsp. ibizensis had the highest number of interspersed repeats, including 24 forward and 20 palindromic repeats. Although there were significant differences between the forward repeats (30–273 bp) and palindromic repeats (30–21,479 bp) in length, the size of most of the interspersed repeats was between 30–35 bp (Figure 5C,D).
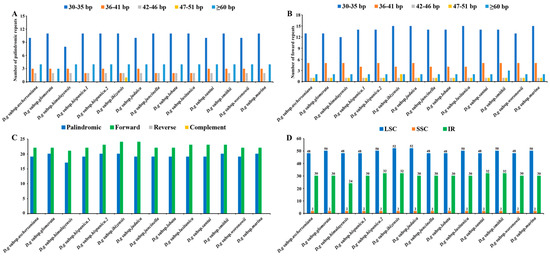
Figure 5.
Analyses of repeated sequences in the cp genomes of 14 D. glomerata subspecies. (A) Number of palindromic repeats; (B) Number of forward repeats by length; (C) Number of different repeats; (D) Numbers of repeats in LSC, SSC and IR regions.
3.5. RNA Editing
We identified 285 RNA editing sites associated with 52 genes in the 14 cp genomes of D. glomerata subspecies (Table S6). Twelve types of RNA editing events were detected, including C to T, A to C, A to T, T to C, T to A, C to G, G to C, G to T, T to G, A to G, G to A, and C to A editing. In addition, all the editing events occurred in the region of proteincoding genes. In terms of the number of RNA editing sites of these genes, rpoB exhibited the most abundant RNA editing (15 sites), followed by rpoC1 and matk (14 sites). ndhF had a value of 13. However, nine genes (psbH, psbK, psbZ, rpl16, rpl22, rps8, ndhG, ndhI, and atpH) had only one RNA editing site. The RNA editing of six genes (atpA–1148, ndhF–62, ndhK–128, petB–611, rps8–3, and rpoA–527) occurred in the cp genomes of all the subspecies. Although most of the editing events caused changes in the amino acids, there were 116 sites that did not alter any amino acids, including stop codons, among the subspecies. A total of 130 sites were edited in the third codon position, 79 sites were in the second codon position, and 74 sites were in the first codon position. Many of the editing events increased the hydrophobicity of amino acids, including conversions from hydrophilic amino acids to hydrophobic amino acids (57), from hydrophilic amino acids to hydrophilic amino acids (83), hydrophobic amino acids to hydrophilic amino acids (20), and hydrophobic amino acids to hydrophobic amino acids (107). Among the 169 edited amino acid sites, the largest proportion of changes was from Ser to Leu (13 sites), followed by Ser to Pro (8 sites), and Pro to Leu (5 sites). RNA editing in D. glomerata subsp. glomerata and D. glomerata subsp. himalayensis was completely consistent and contained 71 RNA editing sites that were involved in 27 genes. On the other hand, RNA editing of D. glomerata subsp. ibizensis and D. glomerata subsp. hispanica.1 was different in many cases from those of the other subspecies, and 77 to 78 editing sites were identified in 34 genes of these two subspecies.
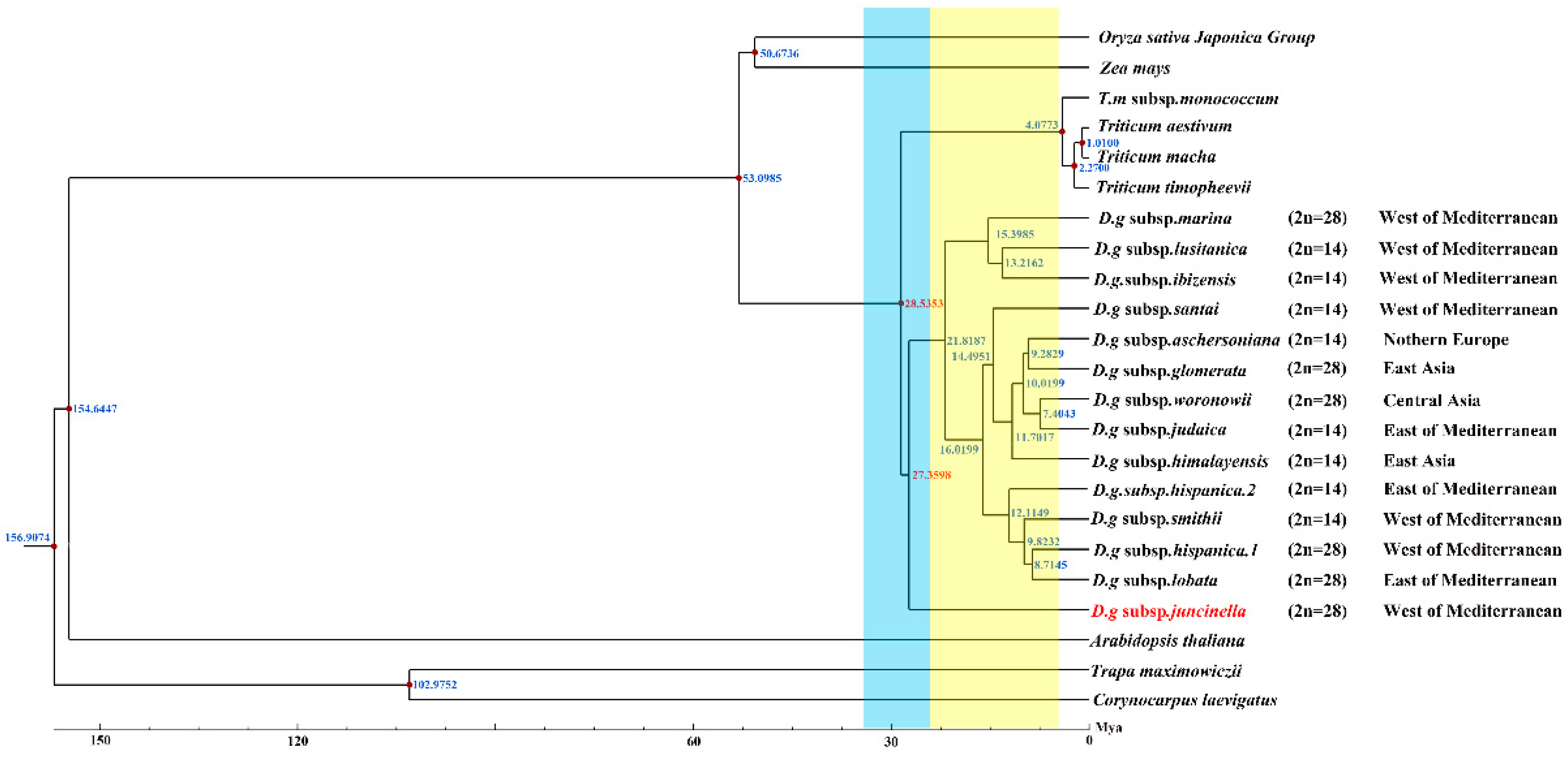
3.6. Phylogenetic Analyses
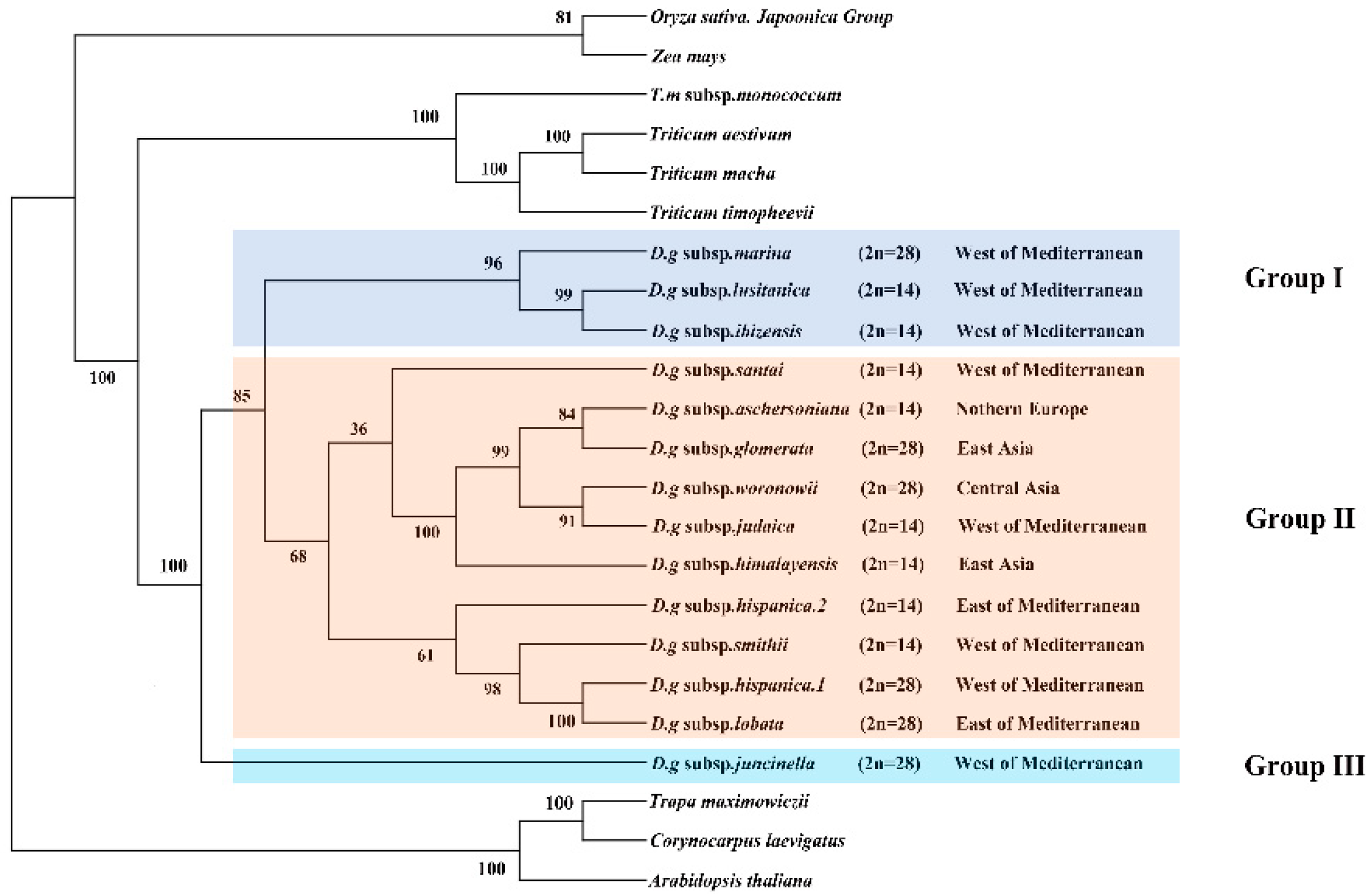
The clade composed of four Triticum species was the sister clade to the D. glomerata species and had BS of 100% (Figure 6). The Dactylis species was strongly supported as a monophyletic group (bootstrap support (BS) = 100%). These 14 subspecies of D. glomerata were divided into three groups. Group I contained three subspecies, group II contained 11 materials of 10 subspecies, and group III contained one subspecies. Although a few nodes still had lower BS values, most of the nodes of the ML tree had high BS values (> 80%). In Group II, it was clear that four D. glomerata subspecies—the diploid subspecies aschersoniana, the tetraploid subspecies glomerata, the tetraploid subspecies woronowii, and the diploid subspecies judaica–were the sister lineages of the diploid subspecies himalayensis and demonstrated higher BS (100%). The diploid subspecies aschersoniana showed the closest relationship with the tetraploid subspecies glomerata followed by the diploid subspecies judaica and the tetraploid subspecies woronowii. The diploid subspecies judaica was closely related to the tetraploid subspecies woronowii. The tetraploid subspecies marina showed a closer relationship with the evolutionary branches composed of the diploid subspecies lusitanica and ibizensis. The tetraploid subspecies lobata and hispanica.1 are sister species, with BS of 100%.
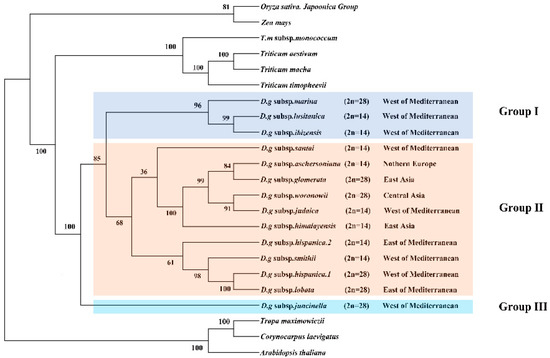
Figure 6.
Phylogeny of Dactylis species inferred from complete chloroplast genome dataset. Phylogenetic tree constructed from D. glomerata subspecies and nine crop complete cp genome sequences using maximum likelihood (ML) method. Bootstrap values are marked above the branches. Arabidopsis, Corynocarpus and Trapa are used as the outgroups.
3.7. Divergence Time
Serial divergence times from the crop plants to D. glomerata were estimated at 53.0985 to 29.5353 Mya (Figure 7). The divergence of the four Triticum species (T. aestivum, T. macha, T. timopheevii, and T. monococcum subsp. monococcum) lagged far behind that of Dactylis glomerata, which shared a common ancestor at 28.5353 Mya. Our study revealed climate change might have been a driver for the divergence of D. glomerata. Based on our results, D. glomerata originated in the early Oligocene era (28.5383 Mya) and gradually began to diversify after the mid–Miocene era (15.3985–7.4043 Mya). The divergence times of the tetraploid subspecies marina and the diploid subspecies santi, lusitanica and ibizensis (15.3985 to 13.2162 Mya), all of which originated from the west of the Mediterranean, were much earlier than those of the other remaining subspecies. Moreover, the divergence event of the diploid subspecies himalayensis, the diploid subspecies aschersoniana, the diploid subspecies judaica, and the tetraploid subspecies glomerata and woronowii occurred in 11.7017 Mya.
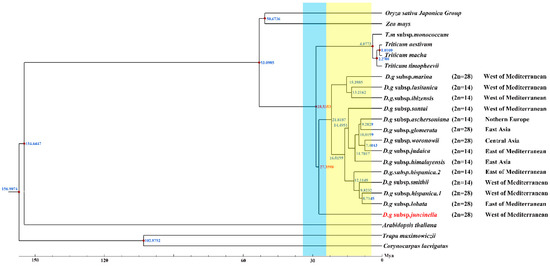
Figure 7.
Divergence time of D. glomerata subspecies. Molecular dating of 14 Dactylis subspecies based on the cp genome sequences in cp genomes. Blue bar represents Oligocene period and yellow bar represents Miocene period.
4. Discussion
4.1. The Features of Dactylis glomerata Subspecies Cp Genomes
In this study, we assembled and compared the characteristics of the cp genomes of 14 subspecies of D. glomerata, with sizes within the range of angiosperms [35]. In agreement with many higher plants, the cp genomes of all the D. glomerata subspecies were highly conserved and displayed a typical quadripartite molecular structure [36,37]. Upon comparing the cp genomes from 14 subspecies of D. glomerata, we found that the genome structures were relatively conserved, with no rearrangement occurring in gene organization, but there were some very significant differences in terms of size among the cp genomes of the analyzed D. glomerata subspecies. The overall genome, LSC, SSC, and IR regions of the D. glomerata subsp. hispanica.1 and the D. glomerata subsp. lobata were shorter than those of the others, which might have been caused by the contraction and expansion of the IR and SSC boundary regions [37,38]. As such, the existing differences may provide insights into the unique differences defining Gramineae species and subspecies [39,40]. Though there were some differences in cp, the genome size among the 14 subspecies, the total GC content and the number of genes were similar, partially reflecting the conservation of the cp genome in angiosperms. In terms of the GC content of the cp genomes of the 14 subspecies of D. glomerata, there was higher GC content in the IR regions than in the LSC and SSC regions, and this could be the result of there being four rRNA genes in the IRs [41]. A high GC content has always been conducive to the stability of the genome structure, making mutation difficult [42]. Thus, the IR was the most conserved region in the cp genome of the 14 subspecies of D. glomerata.
The cp genomes of most angiosperms contain 74 protein–coding genes in their cp genomes; however, five other genes were found among certain species, with variation also being observed in the D. glomerata subspecies [43]. Interestingly, a translation initiation factor, infA, has been independently lost multiple times during the evolution of different land plants. However, it appears in all the subspecies in this study [43]. Thus, we deduced that the presence of infA is an ancestral condition in these subspecies. In addition, a gene with a high frequency of absence from the 14 D. glomerata subspecies was ycf15. The function of ycf15 has been a concern in previous studies–its potential as a protein–coding gene in the cp genome of angiosperms has been questioned due to its high frequency as a pseudogene [44,45,46]. It is estimated that the ycf15 gene has completely disappeared from about 29 terrestrial plant lineages [47,48,49,50,51,52,53,54]. However, comparative analysis revealed that this gene was found in the D. glomerata subsp. hispanica.1 but that it was not present in other listed Dactylis subspecies. Based on these findings, parallel losses of particular genes have occurred over the course of Dactylis evolution. In this study, this finding increased our interest in studying the function and evolution of ycf15 in angiosperms in more detail. Thus, the ycf15 loss event may provide useful information for a more in–depth study of the evolutionary history of Dactylis or of Gramineae species.
4.2. Divergence of Active Regions
In order to explain the level of genome divergence, sequence identity among cp DNAs was detected. Though there was a high degree of similarity at the cp genomic scale, highly divergent regions were found, which included nine non–coding regions (rps19–psbA, psbM–petN, trnG–UCC–trnT–GGU, psaA–ycf3, rbcL–psal, psbE–petL, rps12–trnV–GAC, trnV–GAC–rps12 and rpl2–rpl23), which may be highly useful for further studies of phylogenetic relationships, the identification of species, and population genetics. In general, the cp genomes of the 14 subspecies of D. glomerata showed lower divergence levels in their coding regions and in IRs than in their non–coding regions and SSC regions, and the IR regions were the most conserved. The higher GC content in IRs might partially explain the divergence of the conserved nature of the IR and SC regions [42].
IR is an important indicator for measuring the structural stability of cp genomes. Generally, a high number of rearrangements are detected in cp genomes lacking an IR, and these rearrangement events have also been reported in various terrestrial plant lineages [55]. However, the comparative analysis indicated that no rearrangement events took place in the cp genomes of the 14 subspecies of D. glomerata with IRs. The presence of an IR could prevent the occurrence of rearrangement events, resulting in a lower species rearrangement rate, which might account for the absence of rearrangement events detected in the 14 subspecies of D. glomerata [56]. In addition, large and complex repeat sequences in cp genomes are associated with genome rearrangement and stabilization; they provide important information for understanding the evolutionary history of plant species [57,58,59]. In this study, about 38 to 44 repeats in the cp genomes of the 14 subspecies of D. glomerata were detected, a relatively small number [60]. The size of the repeats was studied in the cp genomes that were sequenced and compared–we found that most of the repeats ranged in size from 30 to 35 bp. Although almost all the repeats were not large repeats (>100 bp), the repeats in Dactylis were more abundant than those in other angiosperm cp genomes, ranging between 7 and 13 [61,62,63]. Given the correlation between repeat sequences and rearrangements, high rearrangement rates are likely to be observed in cp genomes with a high frequency of large repeat sequences (>100 bp) [64]. However, no rearrangement events were found to have occurred in our 14 subspecies of D. glomerata, which might be due to a lack of large repeat sequences. It is, therefore, not surprising that rearrangement events were not observed in the 14 D. glomerata subspecies, which explains the relatively higher degree of stability and conservation among the different Dactylis subspecies.
4.3. IR Contraction and Expansion
Although the IR is the most conserved region in the cp genome, the border contraction and expansion of IRs are regarded as common evolutionary events in plants [65]. Overall, there was a close association between the IR length and genome size. For instance, the D. glomerata subsp. lusitanica possessed the largest cp genome (134,993 bp) and had a relatively long IR (21,472 bp), whereas the D. glomerata subsp. lobata and D. glomerata subsp. hispanica.1 had the smallest cp genomes (134,375–134,376 bp) and the shortest IRs (21,236 bp). Analysis of the contraction and expansion of IRs from 14 subspecies of D. glomerata revealed that the gene distribution at the boundary of the four regions of the cp genome followed a similar rule. However, relatively independent characteristics were observed in the microstructure, such as the locations of ndhH, ndhF and rps15. In Gramineae, ndhH exists near both ends of the SSC region and can extend to the IRs, and it plays an important role in the stability of the IR/SSC junction [66]. As a typical cp genome structure, ndhH extends into the IRs in Dactylis, which may represent an ancestral symplesiomorphy in Gramineae [67]. Longer extension of ndhH into IRa unique to the monophyletic D glomerata subsp. lobata and subsp. hispanica.1 cluster among other subspecies indicates that the genome size variation of the 14 subspecies of D. glomerata was mainly due to border shifts and variation in IRs [68]. Thus, we speculated that ndhH might play an important role in the cp evolution within Dactylis. These variations broaden our knowledge of the evolution and genomic structure of the cp genome of Dactylis.
4.4. Characterization of SSRs
SSRs are considered the results of slipped strand mispairing during DNA replication, which is usually observed in plant cp genomes [69]. The SSRs of plant cp genomes have been widely used for molecular markers for studying species genetic variations due to a high polymorphism rate at the species level [70]. In total, six types of SSRs were identified, and similar numbers of SSRs were detected in the 14 subspecies of D. glomerata. These cpSSRs appeared more frequently in the LSC region than in the SSC and IR regions, consistent with results of other Gramineae species [71]. Among the repeat units that were identified, the mononucleotide repeat (A/T) unit was the most abundant repeat unit, of which A or T repeats accounted for the majority. Our results support the concept that the SSRs in cp genomes are typically composed of short polyadenine (polyA) or polythymine (polyT) repeats and rarely contain tandem guanine (G) or cytosine (C) repeats [72]. Among these SSRs, hexanucleotide repeats showed different distribution patterns. Hexanucleotide repeats were the most common types in all the Oryza species and in certain Phalaris arundinacea [60,73], while hexanucleotide repeats were species–specific in the present study. An assessment of the SSR categories indicated that hexanucleotide repeats only existed in D. glomerata subsp. santai. Since the chloroplast is highly conserved in an–giosperms, cpSSRs can be transferred between species and genera [66]. So, the SSRs identified in the cp genome of Dactylis could be used as useful molecular markers to identify these D. glomerata subspecies and related species in future studies.
4.5. RNA Editing
As a crucial post–transcriptional RNA modification process, RNA editing appears in almost all land plants [74]. In the cp genomes of 14 subspecies of D. glomerata, most of the RNA editing events predicted were of C to T. In these 14 subspecies, the amino acid transformation from serine to leucine was the most common type. The dominance of hydrophilic serine to hydrophobic leucine substitution among non–synonymous RNA editing suggests the evolutionary conservation of RNA editing [75]. The RNA editing sites are often detected in the first or second base of codons, causing the conversion of hydrophilic amino acids to hydrophobic amino acids [76,77]. This transformation increases the hydrophobicity of proteins and enhances their stability [78]. In our present study, approximately 57 of the amino acid changes were from hydrophilicity to hydrophobicity, and the increase in amino acid hydrophobicity might facilitate the formation of core residues in proteins [79]. Thus, the structure formed by hydrophobic mutations in the protein kernel is more stable than that formed by hydrophilic mutations, which may ultimately affect the secondary structure and function of proteins and expand their genetic information [75,80].
However, abundant RNA editing sites were identified in the third codon among D. glomerata subspecies, which were consistent with some Gramineae plants [81,82]. The chloroplast RNA editing of D. glomerata subsp. ibizensis and D. glomerata subsp. hispanica.1 varied more than those of the other D. glomerata subspecies, which may be due to changes in the environmental adaptability or species specificity during long–term evolution. Thus, some specific RNA editing sites were also identified in some of the subspecies, which provided useful information for the origin and evolution of the Dactylis genus based on in–sights from chloroplast RNA editing. To our knowledge, this is the first study to report RNA editing among D. glomerata subspecies, elaborating the evolution of 14 subspecies of D. glomerata with a focus on RNA editing, and laying the foundation for further studies of RNA editing mechanisms in D. glomerata and other types of plants.
4.6. Phylogeny Analysis and Divergence Time
Although the phylogenetic relationships among Dactylis taxa have already been extensively studied using morphological, cytological, isozyme, phenolic flavonoid, and molecular techniques, the classification of the Dactylis genus remains controversial [2,9,83,84,85]. In the present study, the phylogenetic tree, based on the cp genome, contains subspecies with different ploidy levels, and the high bootstrap support shows the capacity of complete cp genomes to enhance the phylogenetic resolutions during the evolution of Dactylis, which might represent useful information on the origin and evolution of Dactylis [86,87]. Dactylis is strongly supported as a monophyletic group and as the sister group of clades consisting of four species of Triticeae. We estimated that Dactylis and four Triticeae species diverged at 28.5353 Mya, which reinforces previous findings using genomics data (17.5–29.6 Mya) [82]. Diversification events in the lineages of Dactylis are primarily predicted to have gradually begun after the mid–Miocene era (15.3985–7.4043 Mya) [88]. It is suggested that there were huge climate changes in the climate after the mid–Miocene era, with the late–Miocene era being colder than the early Oligocene era [88]. This global cooling led to the contraction of tropical forests to lower latitudes, resulting in many open habitats, which presented opportunities for Gramineae plants [89]. As a type of cool grass, the adaptive radiation and diversity of D. glomerata subspecies were probably driven by climate change [82,90]. The tetraploid subspecies marina and the diploid subspecies santai, lusitanica and ibizensis from the western Mediterranean were the species observed during the early successive divergence of Dactylis, suggesting that the western Mediterranean might have been a diverse phylogenetic center in the past.
This study showed that most phylogenetic clades underwent adaptive radiation of the diploid subspecies followed by the tetraploid species. Obviously, most diploids have a much earlier origin than tetraploids, which is also supported by previous conclusions from studies on flavonoid variability [83,84,87]. Notably, the tetraploid subspecies marina clustered with the diploid subspecies lusitanica and ibizensis, indicating that one of these diploid subspecies, or their common ancestor, could be a parental species of tetraploid subspecies. The morphology of these subspecies also supports this hypothesis since the tetraploid subspecies marina possesses characteristic papillose epidermal cells that are very similar to those found in the diploid subspecies ibizensis, indicating that they might be derived from a common ancestor [8]. It has previously been suggested that the tetraploid subspecies glomerata may have evolved from the hybridization of aschersoniana (2n = 14) and woronowii (2n = 48) [83]. We found that it formed a clade with the diploid subspecies aschersoniana in the cp genome tree, supporting this diploid subspecies as one of its parents. Moreover, our results revealed that the tetraploid subspecies woronowii and the diploid subspecies judaica were clustered into a clade, indicating a close relationship between the two. Based on morphological studies, mainly of phenotypic characteristics, Stebbins and Zohary considered the tetraploid subspecies woronowii to be more similar to the xeromorphic diploid subspecies judaica, which has somewhat adapted to a dry summer climate and is considered to be of more recent origin [83]. Thus, these two subspecies may originate from a recent common diploid ancestor. In the present study, the diploid subspecies himalayensis forms a well–supported clade with the diploid subspecies aschersoniana, tetraploid subspecies glomerata and woronowii, and the diploid subspecies judaica, indicating that the five subspecies are closely related. In addition, the tetraploid subspecies woronowii is closely associated with the ancient Eurasian temperate forest flora (including the diploid subspecies himalayensis and aschersoniana) [83]. As Stebbins and Zohary note, the diploid subspecies aschersoniana and the tetraploid subspecies woronowii have similar flavonoid components, whereas the diploid subspecies himalayensis and aschersoniana have more primitive compounds than the tetraploid subspecies woronowii [84]. Obviously, the diploid subspecies himalayensis and aschersoniana originated earlier than the tetraploid subspecies woronowii. In this study, the diploid subspecies himalayensis was found to share a common ancestor with four other subspecies with different ploidy levels at 11.7017 Mya. The diploid subspecies himalayensis revealed similar phylogenetic positions when tetraploids were included, suggesting that the diploid subspecies himalayensis, or its ancestral lineage, may have been a parent of these subspecies. Phylogenomic analysis of the cp genomes will help to uncover the mysteries and controversies surrounding the phylogeny of Dactylis species. However, the sequencing data of the Dactylis subspecies used in this study were limited. Therefore, we recommend the inclusion of cp genome sequences of other subspecies of D. glomerata in future studies to help elucidate the evolution from Dactylis.
5. Conclusions
This is the first report of the whole cp genome of D. glomerata subspecies. The 14 subspecies of D. glomerata showed synteny of gene order and contained similar IR boundary regions in their cp genomes. Moreover, we also obtained important genetic information, including SSRs, repeat sequences, divergent hotspot regions, RNA editing sites, and divergence times associated with the relationships between the D. glomerata species and other Gramineae. Our results may supply insights to resolve taxonomic discrepancies and phylogenetic relationships within the D. glomerata species, accelerating the identification and utilization of forage resources.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes13091621/s1. Table S1: The information of Dactylis glomerata subspecies. Table S2: Summary of complete cp genomes of Dactylis subspecies; Table S3: Gene composition within chloroplast genomes of Dactylis species. Note: Genes *: genes with one intron; Genes **: genes with two introns; Gene (2): Number of copies of multi–copy genes; Genes a: represents the ycf15 is particular in D.g subsp. hispanica.1; Table S4: Location and length of intron–containing genes of Dactylis subspecies cp genomes; Table S5: SSRs of Dactylis subspecies cp genomes; Table S6: Comparison of the RNA editing sites in Dactylis subspecies cp genomes. Note: “a” indicates that it is completely edited; “b” represents unedited.
Author Contributions
X.Z. and L.H. conceived and designed the experiments; Y.J., G.F., Y.X. and Z.H. performed the experiments; Y.J. and G.F. analyzed the data; G.N. and Z.L. contributed reagents/materials/analysis tools; Y.J. and G.F. wrote the paper; and Y.P. and D.L. reviewed and edited the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant numbers NSFC 31872997 and NSFC 32101422, the Sichuan Province’s Science Fund for Distinguished Young Scholars under Grant (2021JDJQ001) and the Sichuan Province’s Science Fund for Intertional Cooperation (2022YFH0058). These funding sources contributed to the design of the study, data collection and analysis, and the writing of the manuscript.
Institutional Review Board Statement
No applicable.
Informed Consent Statement
No applicable.
Data Availability Statements
All the data pertaining to the present study has been included in the tables and/or figures in the manuscript. The authors are pleased to share analyzed/raw data and plant materials upon reasonable request. All the cp genome sequences of this study have been de–posited into the CNGB Sequence Archive (CNSA) of the China National Gene Bank Data Base (CNGBdb) with accession number CNP0002292, https://db.cngb.org/mycngbdb/submissions/pro–ject, accessed on 7 April 2022. The plant materials were provided by the Department of Forage Science, College of Grassland Science and Technology, Sichuan Agricultural University, Chengdu, China.
Acknowledgments
The authors would like to express their gratitude to the following: Yanli Xiong, Dandan Li, and Yi Xiong from Sichuan Agricultural University. The authors gratefully acknowledge grants from the National Natural Science Foundation of China (NSFC 31872997 and NSFC 32101422), the Sichuan Province Science Fund for Distinguished Young Scholars under Grant (2021JDJQ0001), and the Sichuan Province Science Fund for Intertional Cooperation (2022YFH0058) for this research.
Conflicts of Interest
The authors declare that they have no conflict of interests.
References
- Casler, M.D.; Fales, S.L.; Undersander, D.J.; Mcelroy, A.R. Genetic Progress from 40 Years of Orchardgrass Breeding in North America Measured under Hay Management. Can J. Plant Sci. 2001, 81, 713–721. [Google Scholar] [CrossRef]
- Yan, D.; Zhao, X.; Cheng, Y.; Ma, X.; Huang, L.; Zhang, X. Phylogenetic and Diversity Analysis of Dactylis Glomerata Subspecies Using SSR and IT-ISJ Markers. Molecules 2016, 21, 1459. [Google Scholar] [CrossRef] [PubMed]
- Lindner, R.; Garcia, A. Genetic differences between natural populations of diploid and tetraploid Dactylis Glomerata ssp. izcoi. Grass Forage Sci. 2008, 52, 291–297. [Google Scholar] [CrossRef]
- Feng, G.; Xu, X.; Xu, L.; Yang, Z.; Nie, G.; Ma, X.; Huang, L.; Zhang, X. Comparative Transcript Profiling Suggests Distinct Flowering Response of Early- and Late-Flowering Phenotypes in Forage Grass Dactylis glomerata L. J. Plant Growth Regul. 2021, 40, 2124–2138. [Google Scholar] [CrossRef]
- Stewart, A.V.; Ellison, N.W. A molecular phylogenetic framework for cocksfoot (Dactylis glomerata L.) improvement. Crop. Pasture Sci. 2014, 65, 780–786. [Google Scholar] [CrossRef]
- Jones, K.; Borrill, M. Chromosomal status, gene exchange and evolution in Dactylis. Genetica 1962, 32, 296–322. [Google Scholar] [CrossRef]
- Lumaret, R.; Guillerm, J.L.; Delay, J.; Loutfi, A.A.L.; Izco, J.; Jay, M. Polyploidy and habitat differentiation in Dactylis glomerata L. from Galicia (Spain). Oecologia 1987, 73, 436–446. [Google Scholar] [CrossRef]
- Borrill, M. Dactylis marina Borrill, sp. nov., a natural group of related tetraploid forms. J. Linn. Soc. 1961, 56, 431–439. [Google Scholar] [CrossRef]
- Lumaret, R.; Retired, M.B. sCytology, genetics, and evolution in the genus Dactylis. Crit. Rev. Plant Sci. 1988, 7, 55–91. [Google Scholar] [CrossRef]
- Neuhaus, H.E.; Emes, M.J. Nonphotosyanthetic metabolism in plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef]
- Arab, M.M.; Brown, P.J.; Abdollahi-Arpanahi, R.; Sohrabi, S.S.; Askari, H.; Aliniaeifard, S.; Mokhtassi-Bidgoli, A.; Mesgaran, M.B.; Leslie, C.A.; Marrano, A.; et al. Genome-Wide Association Analysis and Pathway Enrichment Provide Insights into the Genetic Basis of Photosynthetic Responses to Drought Stress in Persian Walnut. Hortic. Res. 2022, 9, uhac124. [Google Scholar] [CrossRef] [PubMed]
- Bina, H.; Yousefzadeh, H.; Ali, S.S.; Esmailpour, M. Phylogenetic relationships, molecular taxonomy, biogeography of Betula, with emphasis on phylogenetic position of Iranian populations. Tree Genet. Genomes 2016, 12, 84. [Google Scholar] [CrossRef]
- Jansen, R.K.; Wojciechowski, M.F.; Sanniyasi, E.; Lee, S.-B.; Daniell, H. Complete Plastid Genome Sequence of the Chickpea (Cicer Arietinum) and the Phylogenetic Distribution of Rps12 and ClpP Intron Losses among Legumes (Leguminosae). Mol. Phylogenet. Evol. 2008, 48, 1204–1217. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The Evolution of the Plastid Chromosome in Land Plants: Gene Content, Gene Order, Gene Function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Drouin, G.; Daoud, H.; Xia, J. Relative Rates of Synonymous Substitutions in the Mitochondrial, Chloroplast and Nuclear Genomes of Seed Plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Krak, K.; Vít, P.; Belyayev, A.; Douda, J.; Hreusová, L.; Mandák, B. Allopolyploid Origin of Chenopodium Album s. Str. (Chenopodiaceae): A Molecular and Cytogenetic Insight. PLoS ONE 2016, 11, e0161063. [Google Scholar] [CrossRef] [PubMed]
- Sadat-Hosseini, M.; Bakhtiarizadeh, M.R.; Boroomand, N.; Tohidfar, M.; Vahdati, K. Combining Independent de Novo Assemblies to Optimize Leaf Transcriptome of Persian Walnut. PLoS ONE 2020, 15, e0232005. [Google Scholar] [CrossRef]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2010, 8, 61–65. [Google Scholar] [CrossRef]
- Singh, B.P.; Kumar, A.; Kaur, H.; Singh, H.; Nagpal, A.K. CpGDB: A Comprehensive Database of Chloroplast Genomes. Bioinformation 2020, 16, 171–175. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in Homology Search: HMMER3 and Convergent Evolution of Coiled-Coil Regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Laslett, D. ARAGORN, a Program to Detect TRNA Genes and TmRNA Genes in Nucleotide Sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational Tools for Comparative Genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, A.N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST Databases for the Development and Characterization of Gene-Derived SSR-Markers in Barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Kurtz, S. The Vmatch large scale sequence analysis software. Cent. Bioinform. 2011, 170, 391–392. [Google Scholar]
- Mower, J.P. PREP-Mt: Predictive RNA Editor for Plant Mitochondrial Genes. BMC Bioinform. 2005, 6, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, D.; Michalak, I. RaxmlGUI: A Graphical Front-End for RAxML. Org. Divers. Evol. 2012, 12, 335–337. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative Chloroplast Genomics: Analyses Including New Sequences from the Angiosperms Nuphar Advena and Ranunculus Macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef]
- Li, X.; Zuo, Y.; Zhu, X.; Liao, S.; Ma, J. Complete Chloroplast Genomes and Comparative Analysis of Sequences Evolution among Seven Aristolochia (Aristolochiaceae) Medicinal Species. IJMS 2019, 20, 1045. [Google Scholar] [CrossRef]
- Yao, X.H.; Tang, P.; Li, Z.Z.; Li, D.W.; Liu, Y.F.; Huang, H.W. The First Complete Chloroplast Genome Sequences in Actinidiaceae: Genome Structure and Comparative Analysis. PLoS ONE 2015, 10, e0129347. [Google Scholar]
- 38. Asaf, S.; Khan, A.L.; Khan, A.; Khan, G.; Lee, I.J.; Al-Harrasi, A. Expanded Inverted Repeat Region with Large Scale Inversion in the First Complete Plastid Genome Sequence of Plantago Ovata. Sci. Rep. 2020, 10, 3881. [Google Scholar] [CrossRef]
- Miller, J.T.; Bayer, R.J. Molecular phylogenetics of Acacia (Fabaceae: Mimosoideae) based on the chloroplast MATK coding sequence and flanking TRNK intron spacer regions. Am. J. Bot. 2001, 88, 697–705. [Google Scholar] [CrossRef]
- Scharaschkin, T.; Doyle, J.A. Phylogeny and historical biogeography of Anaxagorea (Annonaceae) using morphology and non-coding chloroplast sequence data. Syst. Bot. 2005, 30, 712–735. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The Complete Chloroplast Genome Sequence of the Medicinal Plant Salvia Miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar]
- Terakami, S.; Matsumura, Y.; Kurita, K.; Kanamori, H.; Katayose, Y.; Yamamoto, T.; Katayama, H. Complete Sequence of the Chloroplast Genome from Pear (Pyrus Pyrifolia): Genome Structure and Comparative Analysis. Tree Genet. Genomes 2012, 8, 841–854. [Google Scholar] [CrossRef]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many Parallel Losses of InfA from Chloroplast DNA during Angiosperm Evolution with Multiple Independent Transfers to the Nucleus. Cochrane Database Syst. Rev. 2001, 13, 645–658. [Google Scholar]
- Goremykin, V.; Hirsch-Ernst, K.I.; Wolfl, S.; Hellwig, F.H. The Chloroplast Genome of the ”Basal” Angiosperm Calycanthus Fertilis-Structural and Phylogenetic Analyses. Plant Syst. Evol. 2003, 242, 119–135. [Google Scholar] [CrossRef]
- Steane, D.A. Complete Nucleotide Sequence of the Chloroplast Genome from the Tasmanian Blue Gum, Eucalyptus Globulus (Myrtaceae). DNA Res. 2005, 12, 215–220. [Google Scholar] [CrossRef]
- Schmitz-Linneweber, C.; Maier, R.M.; Alcaraz, J.-P.; Cottet, A.; Herrmann, R.G.; Mache, R. The Plastid Chromosome of Spinach (Spinacia Oleracea): Complete Nucleotide Sequence and Gene Organization. Plant Mol. Biol. 2001, 45, 307–315. [Google Scholar] [CrossRef]
- Kim, H.T.; Chung, M.G.; Kim, K.-J. Chloroplast Genome Evolution in Early Diverged Leptosporangiate Ferns. Mol. Cells 2014, 37, 372–382. [Google Scholar] [CrossRef]
- Wakasugi, T.; Tsudzuki, J.; Ito, S.; Nakashima, K.; Tsudzuki, T.; Sugiura, M. Loss of All Ndh Genes as Determined by Sequencing the Entire Chloroplast Genome of the Black Pine Pinus Thunbergii. Proc. Natl. Acad. Sci. USA 1994, 91, 9794–9798. [Google Scholar] [CrossRef]
- Wu, C.S.; Lai, Y.T.; Lin, C.P.; Wang, Y.N.; Chaw, S.M. Evolution of reduced and compact chloroplast genomes (cp DNAs) in gnetophytes: Selection toward a lower-cost strategy. Mol. Phylogenet. Evol. 2009, 52, 115–124. [Google Scholar] [CrossRef]
- Tsudzuki, J.; Ito, S.; Tsudzuki, T.; Wakasugi, T.; Sugiura, M. A new gene encoding tRNA (GGG) is present in the chloroplast genome of black pine: A compilation of 32 tRNA genes from black pine chloroplasts. Curr. Genet. 1994, 26, 153–158. [Google Scholar] [CrossRef]
- Li, Z.H.; Qian, Z.H.; Liu, Z.L.; Deng, T.T.; Zu, Y.M.; Zhao, P.; Zhao, G.F. The complete chloroplast genome of Armand pine Pinus armandii, an endemic conifer tree species to China. Mitochondrial. DNA A 2016, 27, 2635–2636. [Google Scholar] [CrossRef] [PubMed]
- Morton, B.R.; Clegg, M.T. A Chloroplast DNA Mutational Hotspot and Gene Conversion in a Noncoding Region near RbcL in the Grass Family (Poaceae). Curr. Genet. 1993, 24, 357–365. [Google Scholar] [CrossRef]
- Ogihara, Y.; Isono, K.; Kojima, T.; Endo, A.; Hanaoka, M.; Shiina, T.; Terachi, T.; Utsugi, S.; Murata, M.; Mori, N.; et al. Structural Features of a Wheat Plastome as Revealed by Complete Sequencing of Chloroplast DNA. Mol. Gene Genom. 2002, 266, 740–746. [Google Scholar] [CrossRef] [PubMed]
- McNeal, J.R.; Kuehl, J.V.; Boore, J.L.; de Pamphilis, C.W. Complete Plastid Genome Sequences Suggest Strong Selection for Retention of Photosynthetic Genes in the Parasitic Plant Genus Cuscuta. BMC Plant Biol. 2007, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Sveinsson, S.; Cronk, Q. Evolutionary Origin of Highly Repetitive Plastid Genomes within the Clover Genus (Trifolium). BMC Evol. Biol. 2014, 14, 228. [Google Scholar] [CrossRef]
- Palmer, J.D.; Osorio, B.; Aldrich, J.; Thompson, W.F. Chloroplast DNA Evolution among Legumes: Loss of a Large Inverted Repeat Occurred Prior to Other Sequence Rearrangements. Curr. Genet. 1987, 11, 275–286. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Chloroplast Evolution: Secondary Symbiogenesis and Multiple Losses. Curr. Biol. 2002, 12, R62–R64. [Google Scholar] [CrossRef]
- Milligan, B.G.; Hampton, J.N.; Palmer, J.D. Dispersed Repeats and Structural Reorganization in Subclover Chloroplast DNA. Mol. Biol. Evol. 1989, 6, 355–368. [Google Scholar]
- Lin, W.-H.; Kussell, E. Evolutionary Pressures on Simple Sequence Repeats in Prokaryotic Coding Regions. Nucleic Acids Res. 2012, 40, 2399–2413. [Google Scholar] [CrossRef]
- Xiong, Y.; Xiong, Y.; Jia, S.; Ma, X. The Complete Chloroplast Genome Sequencing and Comparative Analysis of Reed Canary Grass (Phalaris arundinacea) and Hardinggrass (P. aquatica). Plants 2020, 9, 748. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, X.; Liu, G.; Yin, Y.; Chen, K.; Yun, Q.; Zhao, D.; Al-Mssallem, I.S.; Yu, J. The Complete Chloroplast Genome Sequence of Date Palm (Phoenix Dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef]
- Uthaipaisanwong, P.; Chanprasert, J.; Shearman, J.R.; Sangsrakru, D.; Yoocha, T.; Jomchai, N.; Jantasuriyarat, C.; Tragoonrung, S.; Tangphatsornruang, S. Characterization of the Chloroplast Genome Sequence of Oil Palm (Elaeis Guineensis Jacq.). Gene 2012, 500, 172–180. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; Matzke, A.J.M.; Matzke, M. Complete Sequence and Comparative Analysis of the Chloroplast Genome of Coconut Palm (Cocos Nucifera). PLoS ONE 2013, 8, e74736. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme Reconfiguration of Plastid Genomes in the Angiosperm Family Geraniaceae: Rearrangements, Repeats, and Codon Usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Qian, J.; Li, X.; Sun, Z.; Xu, X.; Chen, S. Complete Chloroplast Genome of Medicinal Plant Lonicera Japonica: Genome Rearrangement, Intron Gain and Loss, and Implications for Phylogenetic Studies. Molecules 2017, 22, 249. [Google Scholar] [CrossRef]
- Davis, J.I.; Soreng, R.J. Migration of Endpoints of Two Genes Relative to Boundaries between Regions of the Plastid Genome in the Grass Family (Poaceae). Am. J. Bot. 2010, 97, 874–892. [Google Scholar] [CrossRef]
- Ma, P.-F.; Liu, Y.-L.; Jin, G.-H.; Liu, J.-X.; Wu, H.; He, J.; Guo, Z.-H.; Li, D.-Z. The Pharus Latifolius Genome Bridges the Gap of Early Grass Evolution. Plant Cell 2021, 33, 846–864. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Guo, X.; Liu, Q.; Wang, K. Complete Chloroplast Genome of Camellia Japonica Genome Structures, Comparative and Phylogenetic Analysis. PLoS ONE 2019, 14, e0216645. [Google Scholar] [CrossRef]
- Levinson, G.; Gutman, G.A. Slipped-Strand Mispairing: A Major Mechanism for DNA Sequence Evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Pandey, S.; Yadav, P.S.; Ansari, W.A.; Pandey, M.; Yang, L.; Singh, B.; Dubey, R.K.; Singh, P.M.; Singh, J. Development of High Conserved Cross-Species Microsatellite Markers from Cucumber Genome and Their Applicability in Genetic Diversity and Comparative Mapping. Sci. Hortic. 2021, 288, 110408. [Google Scholar] [CrossRef]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, K.; Singh, B.; Misra, G.; Singh, N.K. Identification, distribution and comparative analysis of microsatellites in the chloroplast genome of Oryza species. Indian J. Genet. Pl. Br. 2019, 79, 536–544. [Google Scholar] [CrossRef]
- Hiesel, R.; Combettes, B.; Brennicke, A. Evidence for RNA editing in mitochondria of all major groups of land plants except the Bryophyta. Proc. Natl. Acad. Sci. USA 1994, 91, 629–633. [Google Scholar] [CrossRef]
- He, P.; Huang, S.; Xiao, G.; Zhang, Y.; Yu, J. Abundant RNA Editing Sites of Chloroplast Protein-Coding Genes in Ginkgo Biloba and an Evolutionary Pattern Analysis. BMC Plant Biol. 2016, 16, 257. [Google Scholar] [CrossRef]
- Brenner, W.G.; Mader, M.; Müller, N.A.; Hoenicka, H.; Schroeder, H.; Zorn, I.; Fladung, M.; Kersten, B. High Level of Conservation of Mitochondrial RNA Editing Sites Among Four Populus Species. G3-Genes Genom. Genet. 2019, 9, 709–717. [Google Scholar] [CrossRef]
- Odintsova, M.S.; Yurina, N.P. RNA editing in plant chloroplasts and mitochondria. Russ. J. Plant Physl. 2000, 47, 274–284. [Google Scholar]
- Koo, H.J.; Yang, T.-J. RNA Editing May Stabilize Membrane-Embedded Proteins by Increasing Phydrophobicity: A Study of Zanthoxylum Piperitum and Z. Schinifolium Chloroplast NdhG. Gene 2020, 746, 144638. [Google Scholar] [CrossRef]
- Chen, T.C.; Su, Y.Y.; Wu, C.H.; Liu, Y.C.; Huang, C.H.; Chang, C.C. Analysis of mitochondrial genomics and transcriptomics reveal abundant RNA edits and differential editing status in moth orchid, Phalaenopsis aphrodite subsp. formosana. Sci. Hortic. 2020, 267, 109304. [Google Scholar] [CrossRef]
- Halder, R.; Jana, B. Exploring the role of hydrophilic amino acids in unfolding of protein in aqueous ethanol solution. Proteins 2021, 89, 116–125. [Google Scholar] [CrossRef]
- Shi, W.Q.; Deng, P.C.; Li, B.L.; Niu, S.C.; Nie, X.J.; Wang, L.; Baloch, A.W.; Song, W.N. Prediction and Identification of RNA Editing Sites in Chloroplast Transcripts of Brachypodium distachyon. Turk. J. Field Crops 2012, 32, 28–35. [Google Scholar]
- Chen, H.Y.; Deng, L.K.; Jiang, Y.; Lu, P.; Yu, J.N. RNA Editing Sites Exist in Protein-coding genes in the Chloroplast Genome of Cycas taitungensis. J. Integr. Plant Biol. 2011, 53, 961–970. [Google Scholar] [CrossRef]
- Huang, L.K.; Feng, G.Y.; Yan, H.D.; Zhang, Z.R.; Bushman, B.S.; Wang, J.P.; Bombarely, A.; Li, M.Z.; Yang, Z.F.; Nie, G.; et al. Genome assembly provides insights into the genome evolution and flowering regulation of orchardgrass. Plant Biotechnol. J. 2020, 18, 373–388. [Google Scholar] [CrossRef]
- Stebbins, G.L.; Zohary, D. Cytogenetic and evolutionary studies in the genus Dactylis: I: Morphology, distribution and interrelationships of the diploid subspecies. Univ. Calif Berkeley Publ. Bot. 1959, 31, 1. [Google Scholar]
- Fiasson, J.-L.; Ardouin, P.; Jay, M. A Phylogenetic Groundplan of the Specific Complex Dactylis Glomerata. Biochem. Syst. Ecol. 1987, 15, 225–229. [Google Scholar] [CrossRef]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using Plastid Genome-Scale Data to Resolve Enigmatic Relationships among Basal Angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic Analysis of 83 Plastid Genes Further Resolves the Early Diversification of Eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef]
- Stebbins, G.L. Cytogenetics and evolution in the grass family. Am. J. Bot. 1956, 43, 890. [Google Scholar] [CrossRef]
- Beerling, D.J.; Royer, D.L. Convergent Cenozoic CO2 history. Nat. Geosci. 2011, 4, 418–420. [Google Scholar] [CrossRef]
- Prothero, D.R. The late Eocene-Oligocene extinctions. Annu. Rev. Earth Planet Sci. 1994, 22, 145–165. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, X.; Zhao, Y.; Guo, J.; Zhang, T.; Huang, W.; Huang, J.; Hu, Y.; Huang, C.-H.; Ma, H. Phylotranscriptomics Resolves the Phylogeny of Pooideae and Uncovers Factors for Their Adaptive Evolution. Mol. Biol. Evol. 2022, 39, msac026. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).