1. Introduction
The forensic genetics community currently relies on PCR-amplified short tandem repeat (STR) DNA profiles to assess the source of biological evidence associated with criminal activity. Such biological material is often presented as a mixture of DNA from different individuals, necessitating deconvolution of the STR electropherogram results [
1]. An integral part of forensic DNA mixture deconvolution is modeling STR peak amplitude, which varies with DNA input [
2,
3]. Binary DNA interpretation procedures often address this peak height variation through the application of threshold-based heuristics that establish lower bounds for per-contributor intra-locus peak height balance [
4,
5]. In contrast, continuous probabilistic genotyping approaches to interpretation rely on probability distributions to represent variance expectations [
6,
7].
In the case of STRmix™ Probabilistic Genotyping Software, which applies a continuous profile model to forensic DNA mixture data, peak height variation is integrated into STR profile analysis through the application of dynamic variance parameters that are inversely proportional to peak height [
8,
9]. Higher peak heights are accompanied by smaller variance parameters, leading to less allowance for differences between observed peak heights and those modeled by the software. More specifically, the spread of the lognormal distribution of [observed RFU height]/[modeled RFU height] increases with an increase in a variance parameter and decreases with an increase in peak height, as represented mathematically below:
If the peak being modeled is an allele, the relevant peak height is always the height of the allele itself modeled by STRmix v2.8 (i.e., the expected height). Alternatively, if the modeled peak is stutter, the relevant peak height is set by the software user and can be either the expected height of the stutter peak itself or the observed height of the allele giving rise to the stutter. Given a hypothesized set of genotypes, a particular peak may also be composed of signal from both allele and stutter peaks, and in these cases, a shifted lognormal model that combines the relevant allele and stutter lognormals together is utilized [
9].
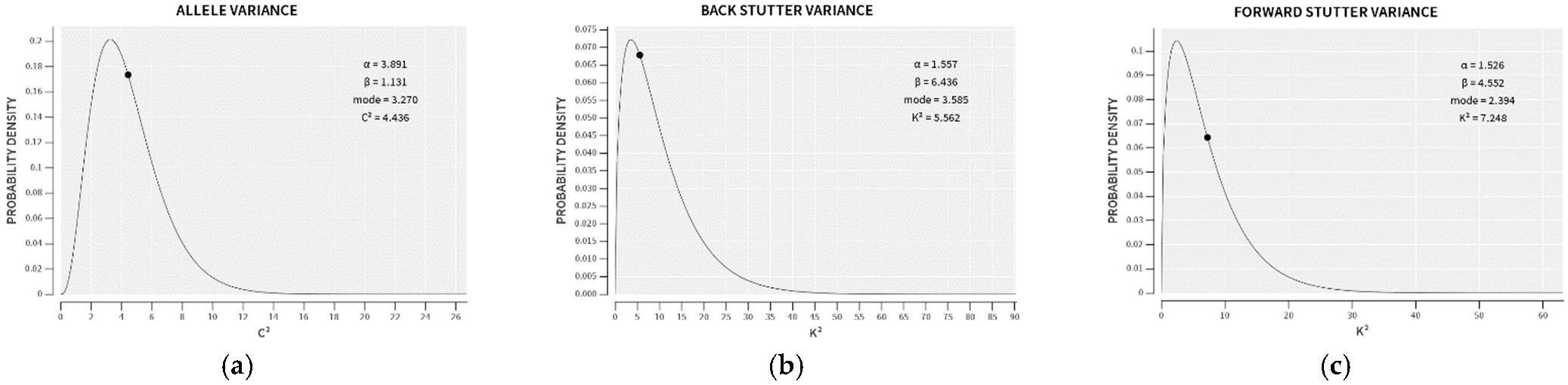
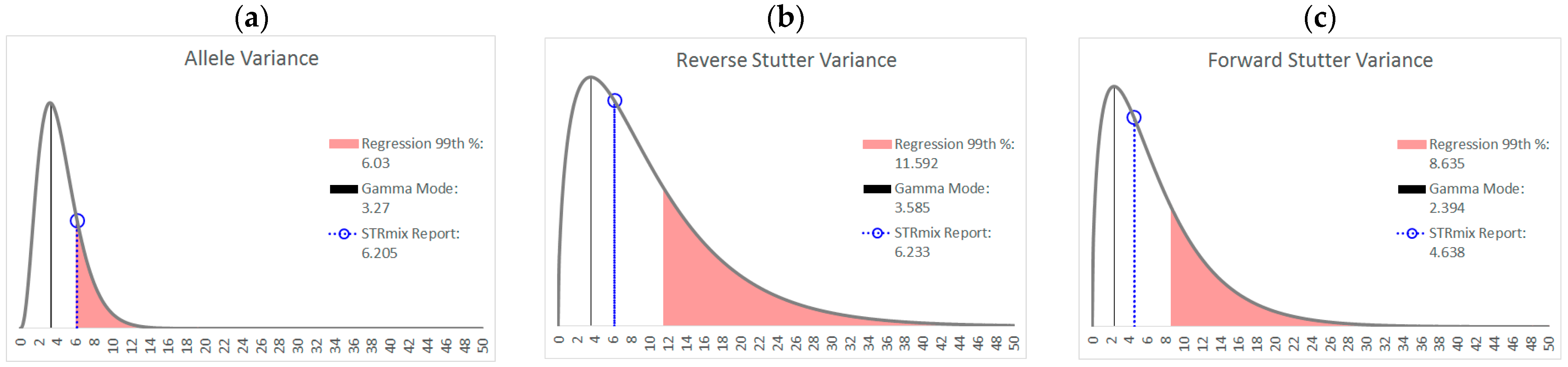
In the preliminary stages of STRmix v2.8 implementation, a large training set of single-source profiles with known types is utilized to construct prior gamma distributions of the variance parameters (see
Figure 1). These prior gamma distributions, created by the Model Maker module in STRmix, are reproduced on every STRmix Interpretation Report, along with the average variance parameters for the interpretation. The creators of STRmix define the genotype probability distributions (GPDs), mixture proportions, and per-locus likelihood ratios (LRs) as primary diagnostics of an interpretation, whereas the average variance parameters are among the secondary STRmix diagnostics that have less well-defined acceptable ranges but provide information about profile modeling efficacy [
10]. Comparison of the average variance parameter for a particular interpretation to the mode of the prior gamma distribution provides a point of reference as to whether greater allowance for variation was required to model the data than was needed for the training set.
However, the comparison of average variance parameters to the prior probability distributions appears to have the same need for threshold calculations that is so central to binary interpretation paradigms. Namely, in order to utilize the average variance parameter values as semi-quantitative diagnostics, it would be important to estimate at what values the average allele and/or stutter variance parameters are far enough away from their prior distribution modes that the STRmix output deserves closer inspection.
Here, we present a systematic examination of STRmix v2.8 allele, reverse stutter (−1 STR repeat), and forward stutter (+1 STR repeat) variance parameters. The goals were to characterize any apparent trends in the variance parameters across a variety of single-source and mixture data, as well as to develop a typical range of parameter values by comparison under standard (i.e., pristine template DNA) and challenging amplification and interpretation conditions. It should be noted that, while both the lower and upper range of variance parameter values are relevant metrics of system performance, STRmix v2.8 has a default lower bound for variance parameters of (0.5) × (prior distribution mode), so we focused exclusively on the upper range in this work.
The value of this study is to increase the usefulness of MCMC (Markov Chain Monte Carlo) summary diagnostics for the forensic DNA community utilizing probabilistic genotyping software. Knowing more about how STRmix variance parameters behave across a large mixture dataset drives empirically supported, reliable assessments of data for forensic case interpretation, addressing related concerns on factor space and likelihood ratio “trustworthiness” as outlined in the recent NISTIR 8351-DRAFT [
11].
4. Discussion
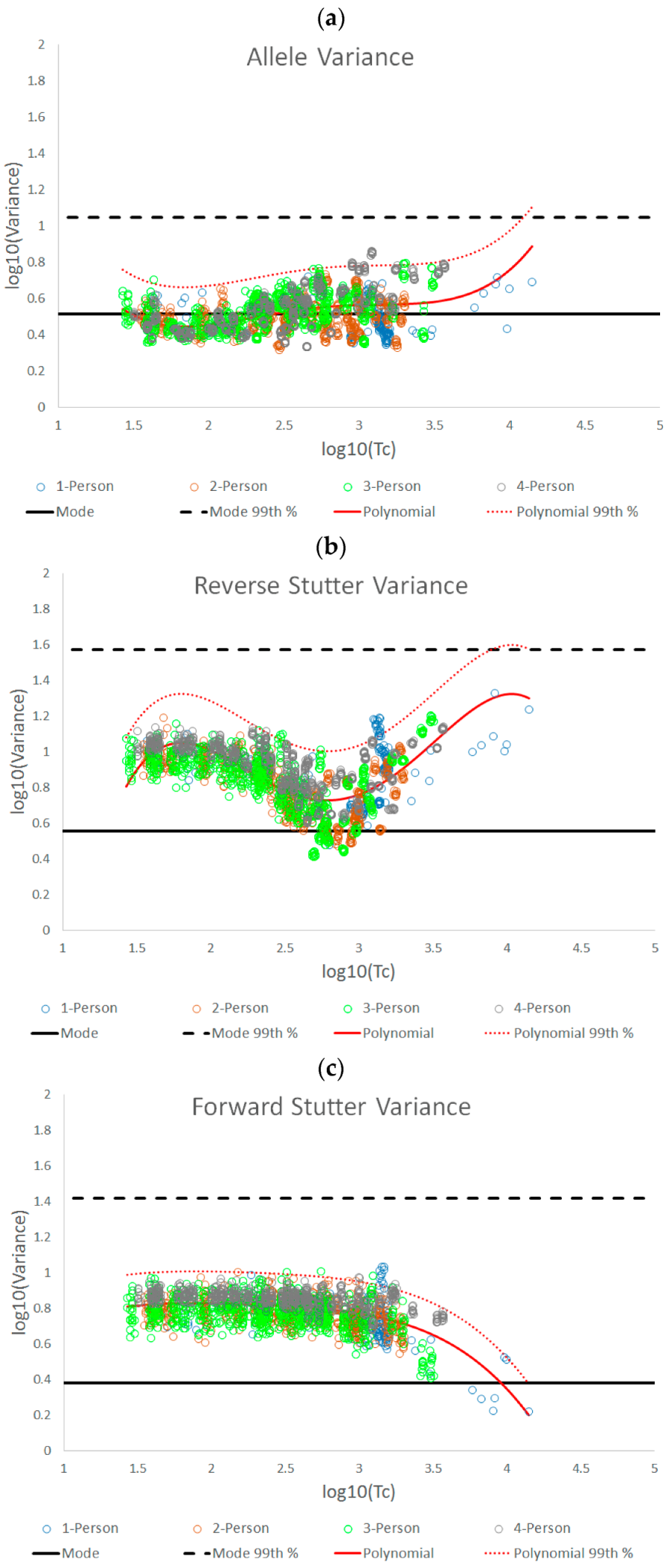
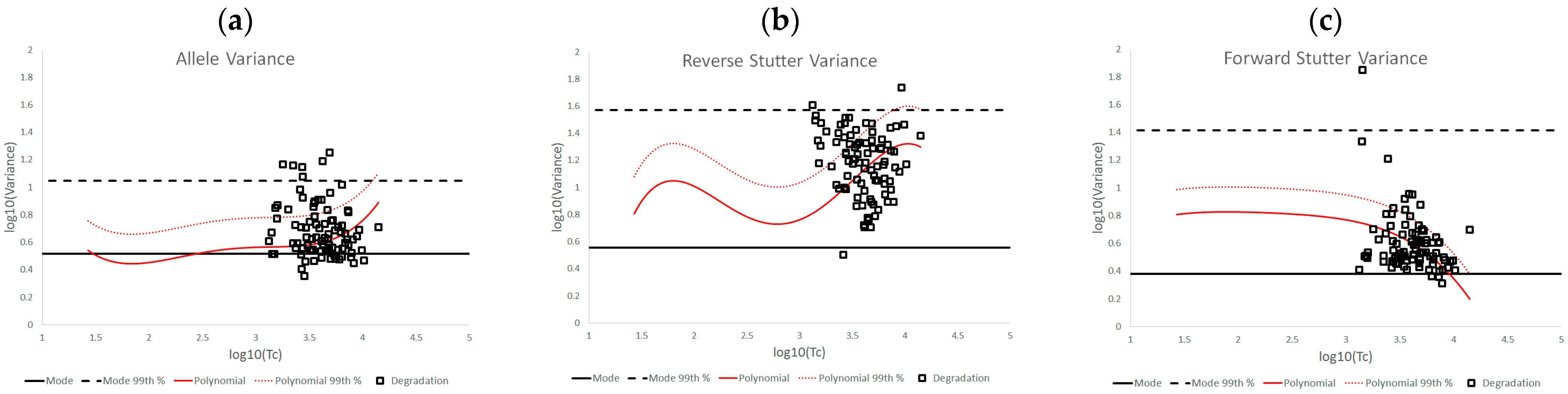
The three STRmix variance parameters we have characterized in this paper fluctuate with increasing template amount, as well as with challenging amplification and/or interpretation conditions. The complexity of the log10(variance parameter) v log10(Tc) plots for the unchallenged data demonstrates the value of establishing an empirically determined working range for STRmix variance parameters instead of assuming that the observed average variance parameter values will always align with the prior gamma distribution.
While this work focused primarily on pragmatic applications of variance parameter data, it is also useful to theorize about what biological or profile modeling factors may have produced the observed patterns in the data. For instance, one might ask why the allele variance parameters for the unchallenged interpretations remained relatively centered on the prior gamma mode across the Tc range, while most of the unchallenged reverse and forward stutter variance parameters (95.28% for reverse stutter and 99.76% for forward stutter) were above the prior mode. These contrasting trends can be attributed to differences in peak detection for alleles, reverse stutters, and forward stutters. While peak height for all detected allelic data is considered during deconvolution, most reverse stutters at low Tc levels will be undetectable, requiring STRmix to model reverse stutter dropout and thus potentially causing the reverse stutter variance to land above the prior mode. The major inflection point of the reverse stutter plot, at a log10(Tc) value of ~2.79, or a Tc of ~617 RFU, corresponds to the approximate point at which stutters begin to be detected. Considering a typical range of reverse stutter ratios to be ~0.05 to 0.1, a Tc of ~617 RFU would equate to reverse stutter peak heights in the range of ~31–62 RFU, which straddles our detection thresholds of 35–71 RFU. Notice that beyond log10(Tc) values of ~2.79, the reverse stutter variance parameters once again move upward, while the allele variance parameters stay relatively flat. A plausible explanation for this trend is that expected stutter peak heights in STRmix are determined by static per-allele stutter ratios and therefore do not have the same degree of model flexibility as expected allele peak heights, which vary with the STRmix template parameter during interpretation. Similar to the reverse stutter variance at log10(Tc) value of ~2.79, the forward stutter variance parameter values begin significantly trending down at a log10(Tc) value of ~3, or a Tc value of ~1000 RFU, which would equate to forward stutter peak heights of ~1–50 RFU (typical forward stutter ratio range of ~0.001 to 0.05), again straddling our detection thresholds and suggesting a change in the modeling fit.
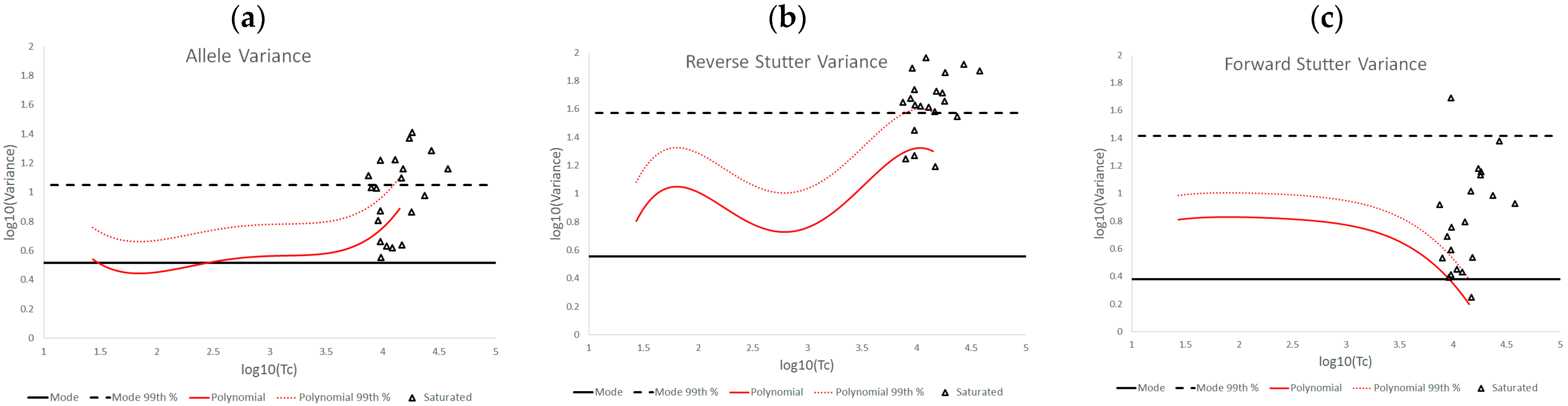
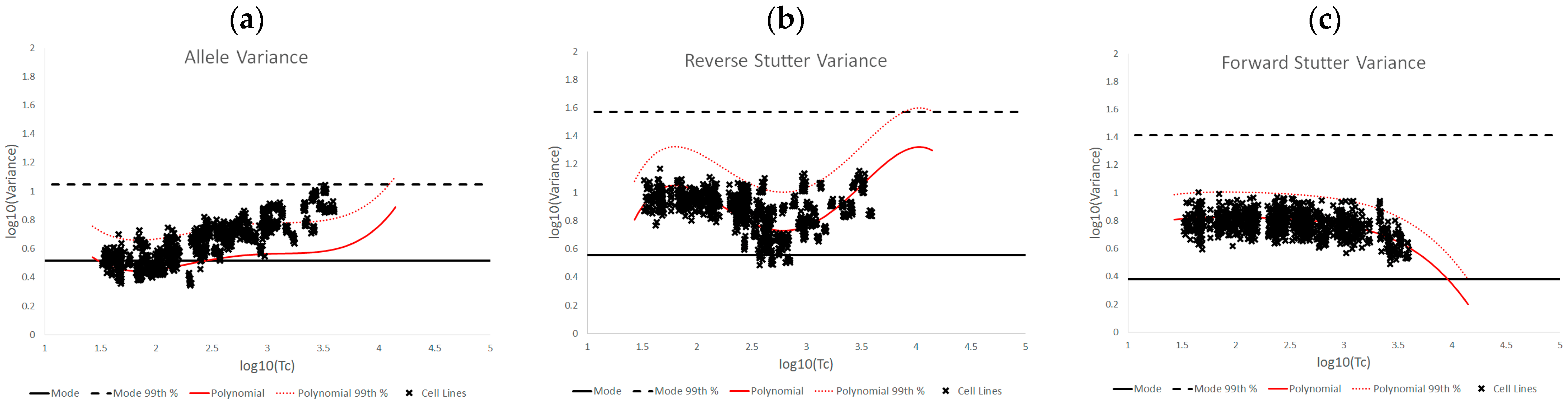
The log
10(variance parameter) v log
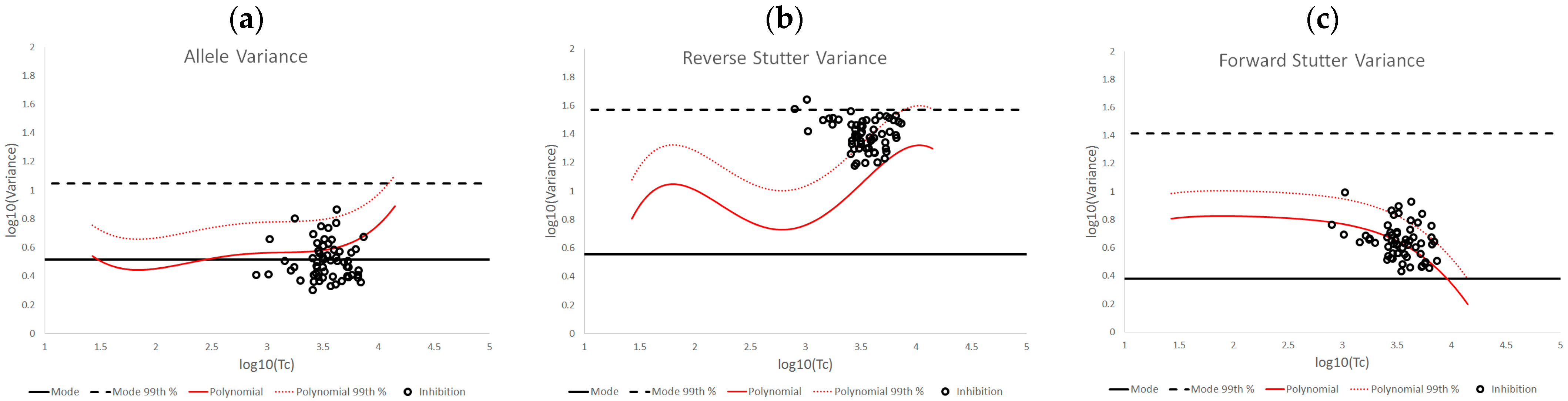
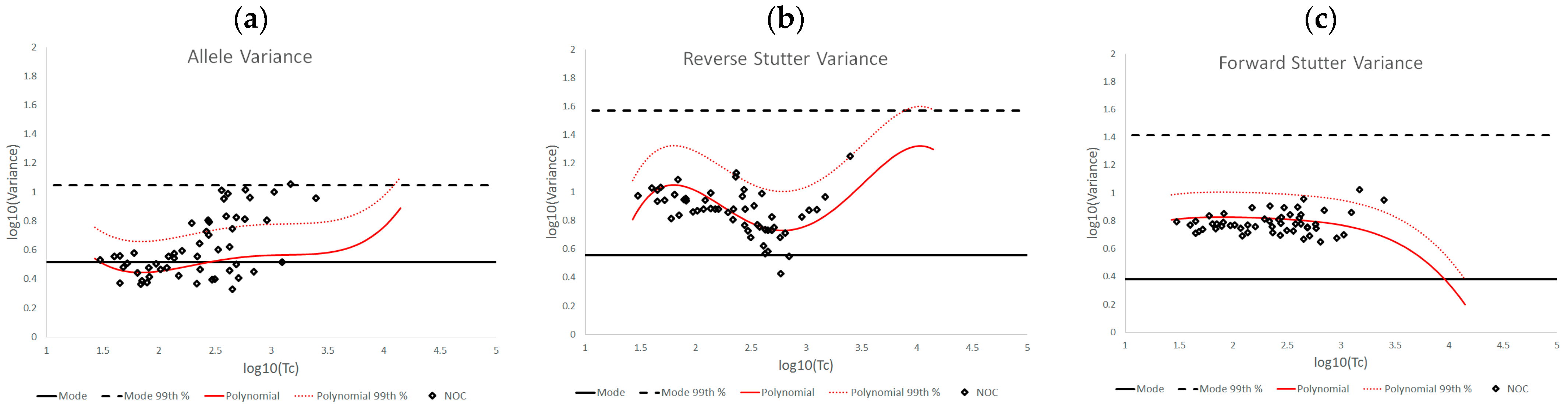
10(Tc) plots of the various challenged datasets also show distinct data patterns that can be associated with biological causes or elements of the STRmix profile model. For the inhibited data set, the most affected of the three variance parameters was reverse stutter, likely because the inhibition required STRmix to model undetected reverse stutter peaks across the profile due to reduced locus yields; in contrast, most of the alleles were still detected at the affected loci. More moderate but significant effects on the reverse stutter variance parameters were observed in the degraded data, which also requires STRmix to model undetected reverse stutter at higher molecular weight loci as peak heights decrease with degradation. However, moderate effects on the allele variance parameters were also observed; these effects are attributable to the difficulty of modeling high levels of degradation, particularly if the value of the exponential decay term in the STRmix degradation model approaches its user-defined ceiling (which occurred with many of the highly degraded mixtures in this set) [
14]. The cell line and underestimated NOC data have similar effects on the variance parameters, in that the allele variance was the most affected of the three. This is a sensible result, given the apparent allele modeling issues with the cell line data and the intralocus allele imbalances that may result from NOC underestimation. Signal saturation, meanwhile, often had a pronounced effect on all three variance parameters. At the peak heights observed in saturated mixtures, tolerance for any peak height deviation from expectation is extremely low, and such deviation is more likely with the loss of linearity between peak height and template, leading to a cascade of effects on the variance parameters. However, not all of the saturated mixtures resulted in elevated variances, because there was variation both in the total number of off-scale peaks detected and the degree of saturation. The more nuanced variance expectations we have presented here can be useful in determining whether the extent of signal saturation observed in a profile has had a discernable effect on its interpretation.
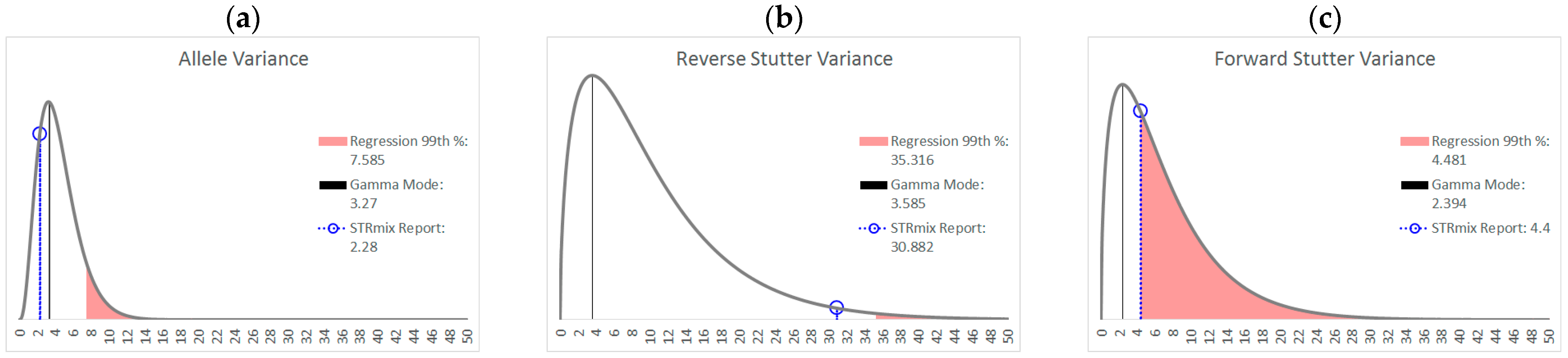
As an example of how a working range for variance parameters might be implemented,
Figure 8 shows the prior gamma distributions for allele, reverse stutter, and forward stutter variance parameters, overlaid with the corresponding parameters for a cell line mixture that resulted in an LR of 0 for the true minor contributor. While the electropherogram for this mixture was unremarkable (see
Supplementary Figure S2), the allele variance parameter was slightly elevated compared to the 99th percentile of our unchallenged log
10(allele variance parameter) v log
10(Tc) regression, which serves as a prompt for closer scrutiny of the interpretation, as well as contributing to an explanation for the aberrant LR result.
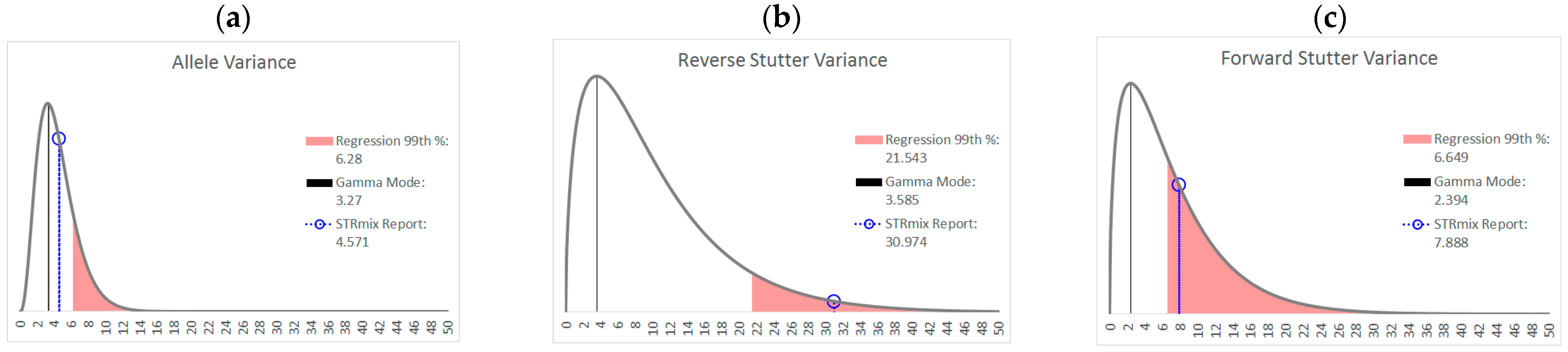
Figure 9 and
Figure 10 are two further examples of how the unchallenged regression data could be applied for routine use in the assessment of STRmix variance parameters from a case result. The interpretations assessed in both figures are from the inhibited mixture data set. In both cases, the allele variance parameter is below the 99th percentile of the unchallenged regression, but in
Figure 9, neither stutter parameter is flagged as high, while in
Figure 10 both are flagged. Notice in particular how similar the reverse stutter parameters are between the two interpretations; despite this similarity, a higher threshold for the reverse stutter variance parameter was applied to the 2-person data because it had a higher Tc than the 3-person data.
Despite the focus in this study on more precisely defining typical ranges for variance parameters, it is important to point out that the observation of a variance parameter outside of the norm does not by itself invalidate a STRmix interpretation; rather, it indicates that more variation than usual was needed for profile modeling. While the final LR is not necessarily a direct measure of how well an interpretation reflects the true contributors’ genotypes, it is notable that the variance outliers of the 3308 interpretations conducted in our study included only one instance of a false exclusion (i.e., an LR of 0 for a true contributor where Hp = true contributor + N-1 unrelated unknown contributors and Hd = N unrelated unknown contributors). The data giving rise to the false exclusion was for the 870 pg 9:1 mixture from the cell line data set featured in
Figure 8. In this case, higher variances, more consistent with the trend in
Figure 7, would have been necessary to capture the allele peak height variation observed in this mixture and avoid the false exclusion. This points to cell line DNA mixtures as potentially inappropriate validation samples for mixture data calibrated to casework-type samples.
While the variance parameter thresholds presented here easily lend themselves to the imposition of binary definitions of “good” and “bad” data, these labels are not appropriate to apply in such a rigid manner. As secondary diagnostics, variances are intended to encourage closer inspection of the input peak data as well as the results of the interpretation, i.e., in this instance, the genotype combinations that STRmix determined to be acceptable explanations of the electropherogram in question. However, despite the utility of secondary diagnostics as indicators of challenged input data, analyst appraisal of the electropherogram data and primary diagnostics can and should be the key measures by which interpretation reliability is assessed.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









