
Estimation of Linkage Disequilibrium, Effective Population Size, and Genetic Parameters of Phenotypic Traits in Dabieshan Cattle

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Genetic Data
2.2. Minor Allele Frequency (MAF)
2.3. Phenotypic Data
2.4. Estimation of LD
2.5. Estimation of Ancestral and Contemporary Ne
2.6. Estimation of Individual’s Genomic Relationship
2.7. Estimation of Genetic Parameter
3. Results
3.1. Genotype Data and Quality Control
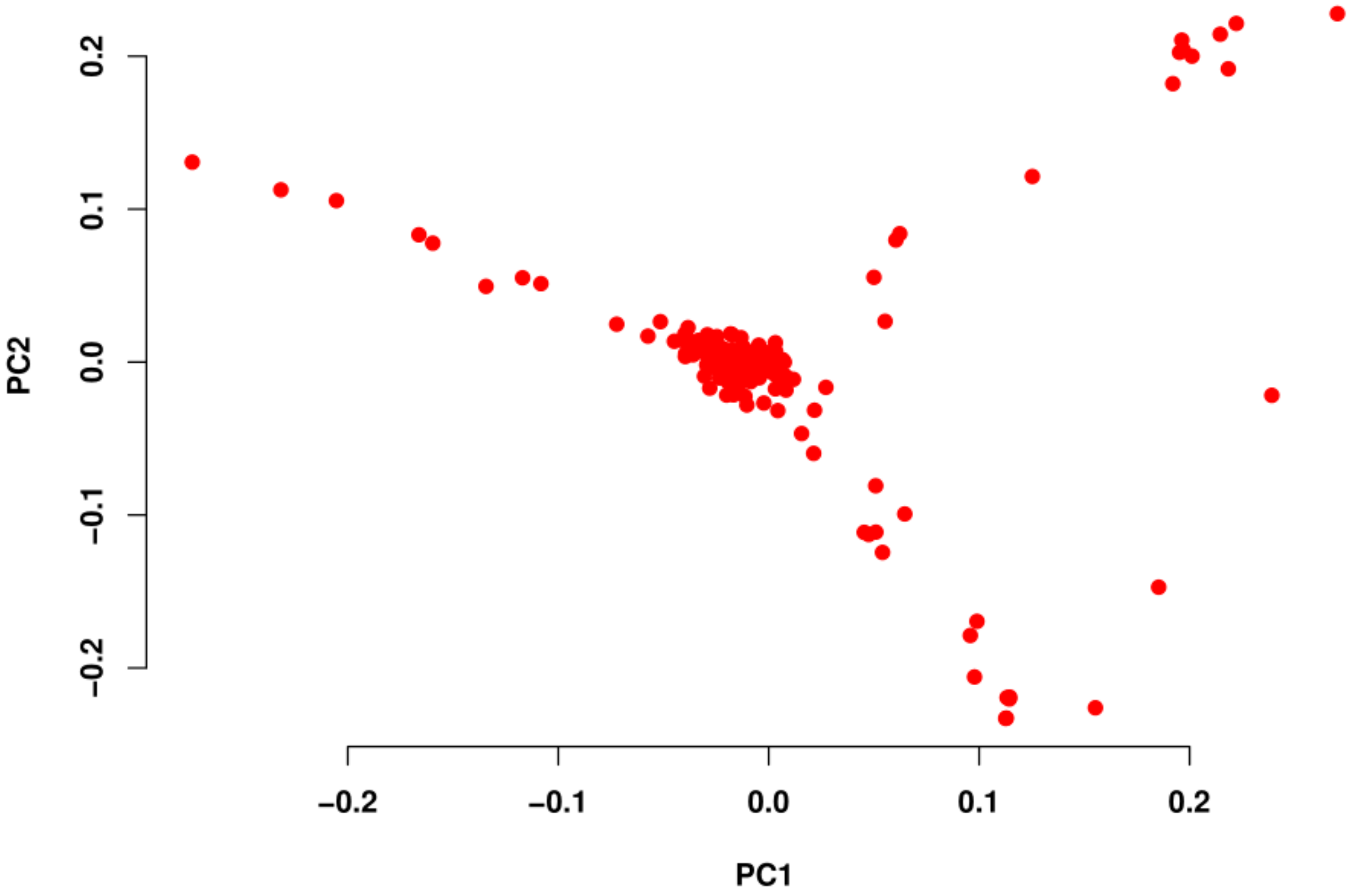
3.2. MAF, GRM, and PCA
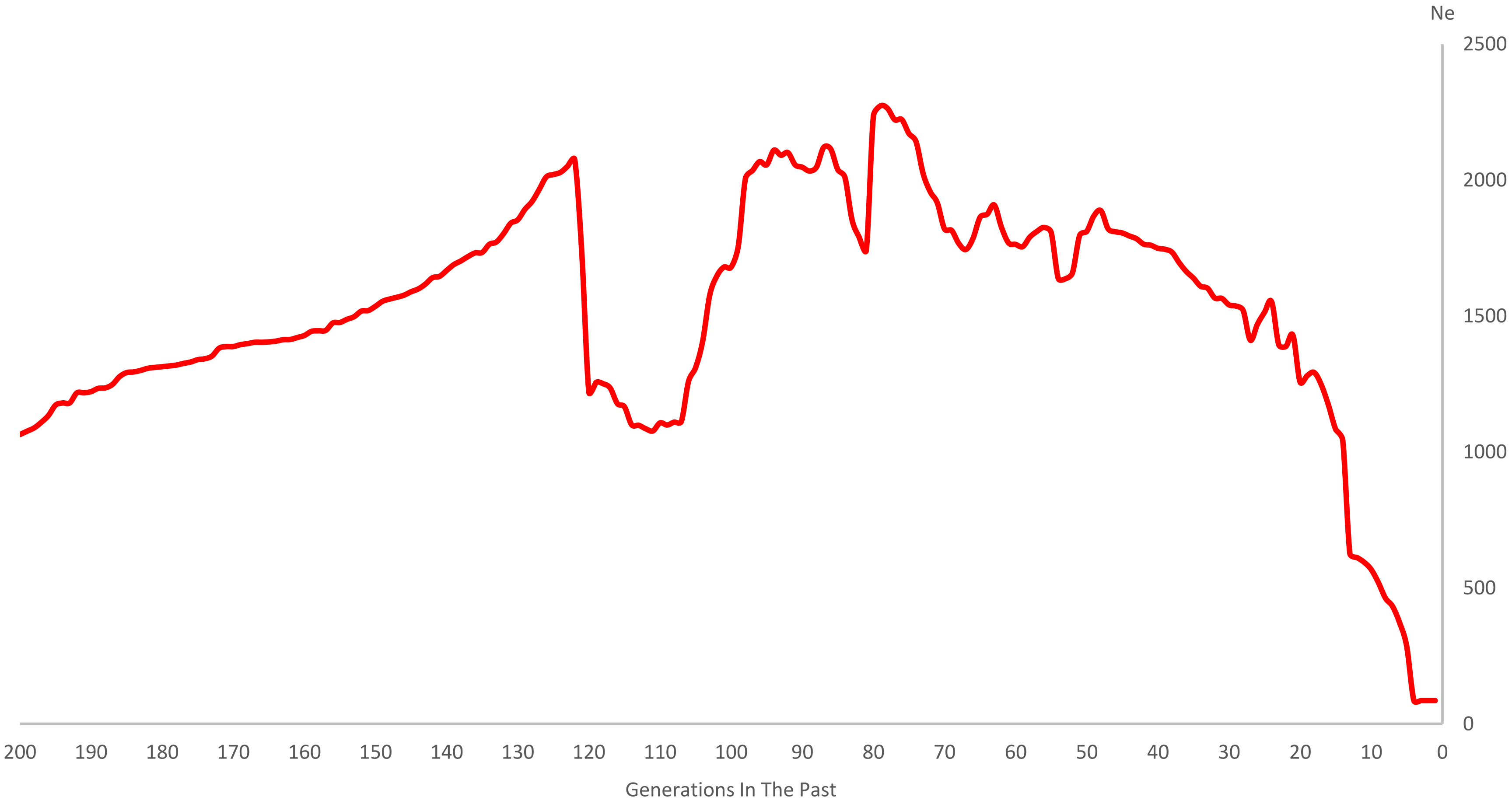
3.3. LD and Ne
3.4. Estimation of Genetic Parameters of Phenotypic Traits
4. Discussion
4.1. MAF, GRM, and PCA
4.2. Extent of LD
4.3. Estimation of Ne
4.4. Estimation of Genetic Parameters of Phenotypic Traits
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, A.R.; Inouye, B.D.; Johnson, M.T.J.; Underwood, N.; Vellend, M. Ecological consequences of genetic diversity. Ecol. Lett. 2008, 11, 609–623. [Google Scholar] [CrossRef] [PubMed]
- China national commission of animal genetic resources. Animal Genetic Resources in China-Bovine; China Agricultural Press: Beijing, China, 2011. [Google Scholar]
- Guan, X.; Zhao, S.; Xiang, W.; Jin, H.; Chen, N.; Lei, C.; Jia, Y.; Xu, L. Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing. Biology 2022, 11, 1327. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhu, B.; Wang, Z.; Xu, L.; Liu, Y.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Zhang, S.; et al. Evaluation of Linkage Disequilibrium, Effective Population Size and Haplotype Block Structure in Chinese Cattle. Animals 2019, 9, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Roos, A.; Hayes, B.J.; Spelman, R.; Goddard, M. Linkage disequilibrium and persistence of phase in Holstein–Friesian, Jersey and Angus cattle. Genetics 2008, 179, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Mrode, R.; Ojango, J.M.K.; Okeyo, A.M.; Mwacharo, J.M. Genomic Selection and Use of Molecular Tools in Breeding Programs for Indigenous and Crossbred Cattle in Developing Countries: Current Status and Future Prospects. Front. Genet. 2019, 9, 694. [Google Scholar] [CrossRef] [Green Version]
- Porto-Neto, L.R.; Kijas, J.W.; Reverter, A. The extent of linkage disequilibrium in beef cattle breeds using high-density SNP genotypes. Genet. Sel. Evol. 2014, 46, 22. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kumar, A.; Mehrotra, A.; Pandey, A.K.; Mishra, B.P.; Dutt, T. Estimation of linkage disequilibrium levels and allele frequency distribution in crossbred Vrindavani cattle using 50K SNP data. PLoS ONE 2021, 16, e0259572. [Google Scholar] [CrossRef]
- Gurgul, A.; Semik, E.; Pawlina, K.; Szmatoła, T.; Jasielczuk, I.; Bugno-Poniewierska, M. The application of genome-wide SNP genotyping methods in studies on livestock genomes. J. Appl. Genet. 2014, 55, 197–208. [Google Scholar] [CrossRef]
- O’Brien, A.M.P.; Mészáros, G.; Utsunomiya, Y.T.; Sonstegard, T.S.; Garcia, J.F.; Van Tassell, C.P.; Carvalheiro, R.; da Silva, M.V.; Sölkner, J. Linkage disequilibrium levels in Bos indicus and Bos taurus cattle using medium and high density SNP chip data and different minor allele frequency distributions. Livest. Sci. 2014, 166, 121–132. [Google Scholar] [CrossRef]
- Leroy, G.; Mary-Huard, T.; Verrier, E.; Danvy, S.; Charvolin, E.; Danchin-Burge, C. Methods to estimate effective population size using pedigree data: Examples in dog, sheep, cattle and horse. Genet. Sel. Evol. 2013, 45, 1–10. [Google Scholar] [CrossRef]
- Palstra, F.P.; Ruzzante, D.E. Genetic estimates of contemporary effective population size: What can they tell us about the importance of genetic stochasticity for wild population persistence? Mol. Ecol. 2008, 17, 3428–3447. [Google Scholar] [CrossRef]
- Crow, J.F.; Kimura, M. An Introduction to Population Genetics Theory. Population 1971, 26, 977. [Google Scholar] [CrossRef]
- Falconer, D.S. Introduction to Quantitative Genetics; Pearson Education India: Noida, India, 1996. [Google Scholar]
- Sørensen, A.; Sørensen, M.; Berg, P. Inbreeding in Danish Dairy Cattle Breeds. J. Dairy Sci. 2005, 88, 1865–1872. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the genomics era—concepts and misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Hill, W.G.; Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 1968, 38, 226–231. [Google Scholar] [CrossRef]
- Santiago, E.; Novo, I.; Pardiñas, A.F.; Saura, M.; Wang, J.; Caballero, A. Recent Demographic History Inferred by High-Resolution Analysis of Linkage Disequilibrium. Mol. Biol. Evol. 2020, 37, 3642–3653. [Google Scholar] [CrossRef]
- Mitchell, M. An Introduction to Genetic Algorithms; MIT Press: Cambridge, MA, USA, 1998. [Google Scholar]
- VanRaden, P. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Xia, X.; Jia, Y.; Dang, R.; Chen, H.; Lei, C. Y-SNPs and Y-STRs Genetic Diversity and Paternal Origin of Dabieshan Cattle. China Cattle Sci. 2018, 44, 4–6. [Google Scholar]
- McKay, S.D.; Schnabel, R.D.; Murdoch, B.M.; Matukumalli, L.K.; Aerts, J.; Coppieters, W.; Crews, D.; Neto, E.D.; Gill, C.A.; Gao, C.; et al. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 2007, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tiezzi, F.; Maltecca, C. Accounting for trait architecture in genomic predictions of US Holstein cattle using a weighted realized relationship matrix. Genet. Sel. Evol. 2015, 47, 24. [Google Scholar] [CrossRef] [PubMed]
- Rokouei, M.; Torshizi, R.V.; Shahrbabak, M.M.; Sargolzaei, M.; Sørensen, A. Monitoring inbreeding trends and inbreeding depression for economically important traits of Holstein cattle in Iran. J. Dairy Sci. 2010, 93, 3294–3302. [Google Scholar] [CrossRef] [PubMed]
- Saatchi, M.; McClure, M.C.; McKay, S.D.; Rolf, M.M.; Kim, J.; Decker, J.E.; Taxis, T.M.; Chapple, R.H.; Ramey, H.R.; Northcutt, S.L.; et al. Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet. Sel. Evol. 2011, 43, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasielczuk, I.; Gurgul, A.; Szmatoła, T.; Semik-Gurgul, E.; Pawlina-Tyszko, K.; Szyndler-Nędza, M.; Blicharski, T.; Szulc, K.; Skrzypczak, E.; Bugno-Poniewierska, M. Comparison of linkage disequilibrium, effective population size and haplotype blocks in Polish Landrace and Polish native pig populations. Livest. Sci. 2020, 231, 103887. [Google Scholar] [CrossRef]
- Lu, D.; Sargolzaei, M.; Kelly, M.; Li, C.; Voort, G.V.; Wang, Z.; Plastow, G.; Moore, S.; Miller, S.P. Linkage disequilibrium in Angus, Charolais, and Crossbred beef cattle. Front. Genet. 2012, 3, 152. [Google Scholar] [CrossRef] [Green Version]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [Green Version]
- Villa-Angulo, R.; Matukumalli, L.K.; Gill, C.; Choi, J.; Van Tassell, C.P.; Grefenstette, J.J. High-resolution haplotype block structure in the cattle genome. BMC Genet. 2009, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rogers, A.R. How Population Growth Affects Linkage Disequilibrium. Genetics 2014, 197, 1329–1341. [Google Scholar] [CrossRef] [Green Version]
- Khatkar, M.S.; Nicholas, F.W.; Collins, A.R.; Zenger, K.R.; Cavanagh, J.A.L.; Barris, W.; Schnabel, R.D.; Taylor, J.F.; Raadsma, H.W. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genom. 2008, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Barbato, M.; Orozco-Terwengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Novo, I.; Santiago, E.; Caballero, A. The estimates of effective population size based on linkage disequilibrium are virtually unaffected by natural selection. PLoS Genet. 2022, 18, e1009764. [Google Scholar] [CrossRef]
- Ge, T.; Holmes, A.J.; Buckner, R.L.; Smoller, J.W.; Sabuncu, M.R. Heritability analysis with repeat measurements and its application to resting-state functional connectivity. Proc. Natl. Acad. Sci. USA 2017, 114, 5521–5526. [Google Scholar] [CrossRef] [Green Version]
- Conley, D.; Siegal, M.L.; Domingue, B.; Harris, K.M.; McQueen, M.B.; Boardman, J.D. Testing the key assumption of heritability estimates based on genome-wide genetic relatedness. J. Hum. Genet. 2014, 59, 342–345. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Hemani, G.; Vinkhuyzen, A.A.; Chen, G.B.; Lee, S.H.; Wray, N.R.; Goddard, M.E.; Yang, J. Statistical Power to Detect Genetic (Co)Variance of Complex Traits Using SNP Data in Unrelated Samples. PLoS Genet. 2014, 10, e1004269. [Google Scholar] [CrossRef] [Green Version]
- Loberg, A.; Dürr, J.W.; Fikse, W.; Jorjani, H.; Crooks, L. Estimates of genetic variance and variance of predicted genetic merits using pedigree or genomic relationship matrices in six Brown Swiss cattle populations for different traits. J. Anim. Breed. Genet. 2015, 132, 376–385. [Google Scholar] [CrossRef]
- Saatchi, M.; Schnabel, R.D.; Rolf, M.M.; Taylor, J.F.; Garrick, D.J. Accuracy of direct genomic breeding values for nationally evaluated traits in US Limousin and Simmental beef cattle. Genet. Sel. Evol. 2012, 44, 38. [Google Scholar] [CrossRef] [Green Version]
- Gunia, M.; Saintilan, R.; Venot, E.; Hozé, C.; Fouilloux, M.N.; Phocas, F. Genomic prediction in French Charolais beef cattle using high-density single nucleotide polymorphism markers1. J. Anim. Sci. 2014, 92, 3258–3269. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype Trait | h2 ± SE | ||
|---|---|---|---|
| Wither Height (WH) | 16.523 | 42.488 | 0.28 ± 0.06 |
| Height at Hip Cross (HHC) | 22.189 | 31.930 | 0.41 ± 0.04 |
| Diagonal Body Length (DBL) | 36.585 | 94.075 | 0.28 ± 0.07 |
| Chest Girth (CG) | 133.103 | 310.574 | 0.30 ± 0.08 |
| Abdomen Circumference (AC) | 86.329 | 530.305 | 0.14 ± 0.06 |
| Waist Angle Width (WAW) | 5.049 | 21.525 | 0.19 ± 0.05 |
| Ischial End Width (IEW) | 1.523 | 11.167 | 0.12 ± 0.09 |
| Shin Circumference (SC) | 2.138 | 1.821 | 0.54 ± 0.07 |
| Body Weight (BW) | 1132.382 | 1928.109 | 0.37 ± 0.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Zhao, S.; Jia, Y.; Xu, L. Estimation of Linkage Disequilibrium, Effective Population Size, and Genetic Parameters of Phenotypic Traits in Dabieshan Cattle. Genes 2023, 14, 107. https://doi.org/10.3390/genes14010107
Jin H, Zhao S, Jia Y, Xu L. Estimation of Linkage Disequilibrium, Effective Population Size, and Genetic Parameters of Phenotypic Traits in Dabieshan Cattle. Genes. 2023; 14(1):107. https://doi.org/10.3390/genes14010107
Chicago/Turabian StyleJin, Hai, Shuanping Zhao, Yutang Jia, and Lei Xu. 2023. "Estimation of Linkage Disequilibrium, Effective Population Size, and Genetic Parameters of Phenotypic Traits in Dabieshan Cattle" Genes 14, no. 1: 107. https://doi.org/10.3390/genes14010107