
Phylogeography of Toona ciliata (Meliaceae) Complex in China Inferred from Cytonuclear Markers

Abstract
:1. Introduction
2. Materials and Methods
2.1. Population Sampling and DNA Extraction
2.2. Primer Screening, Amplification and Sequencing
2.3. Analysis of Genetic Diversity
2.4. Population Genetic Structure
2.5. Population Demography
2.6. Ratio of Pollen to Seed Flow
3. Results
3.1. Haplotype Analysis
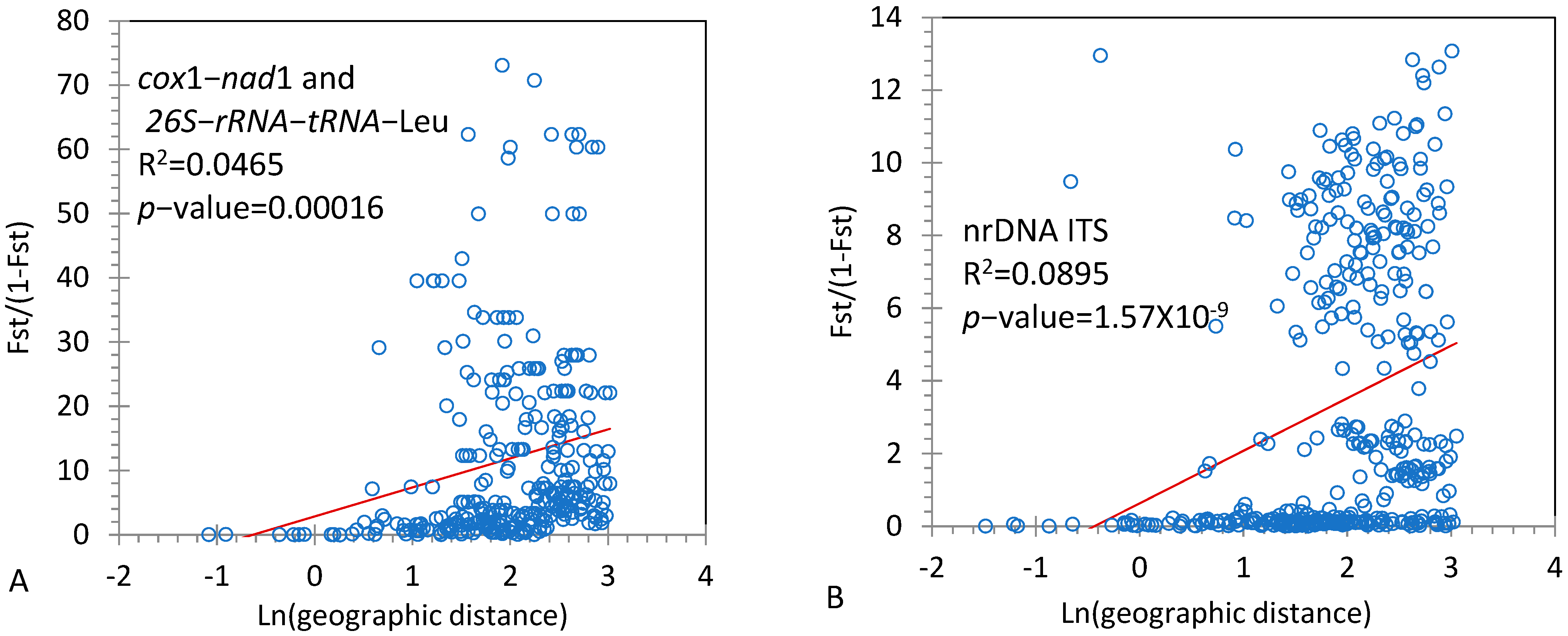
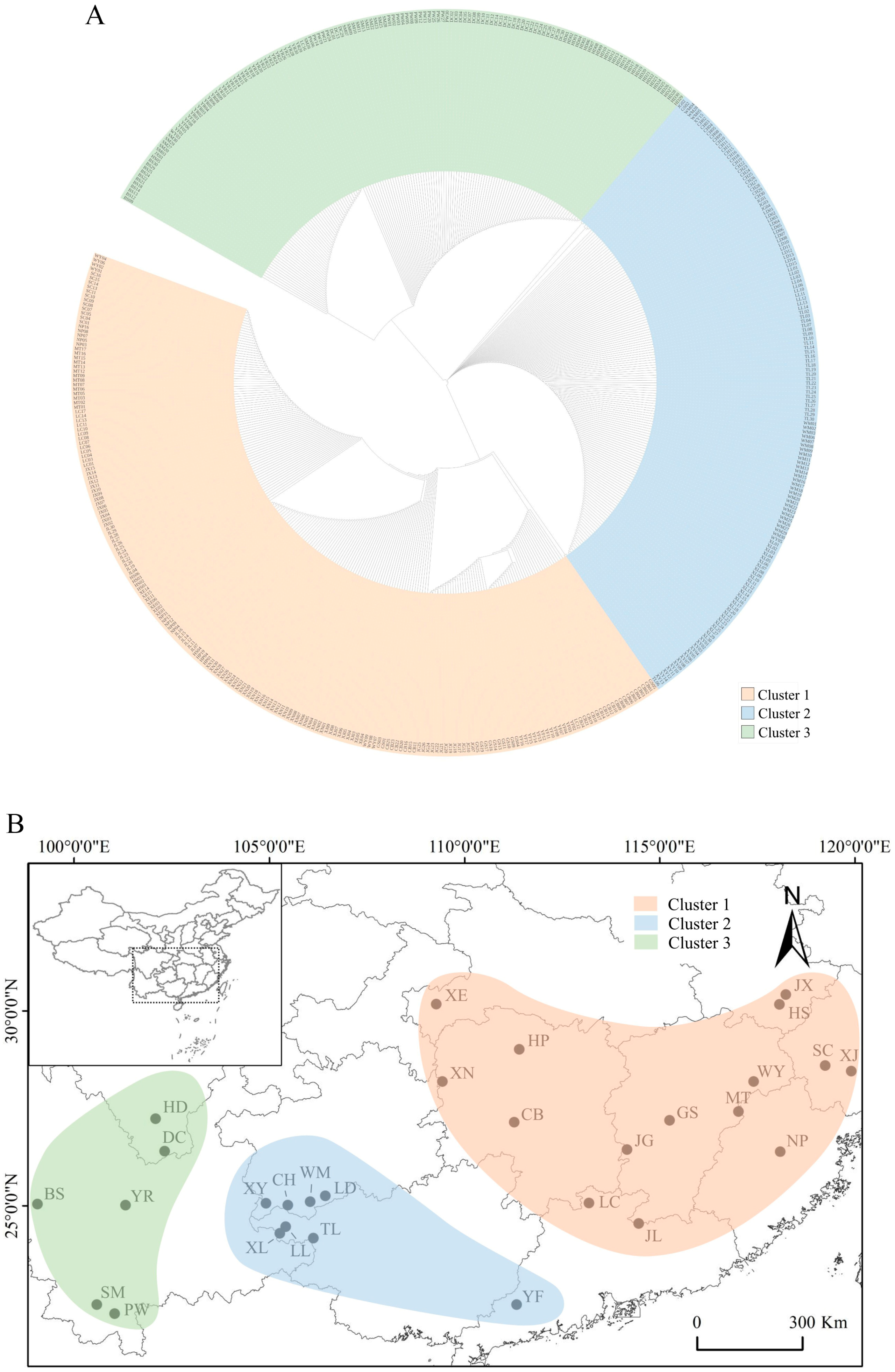
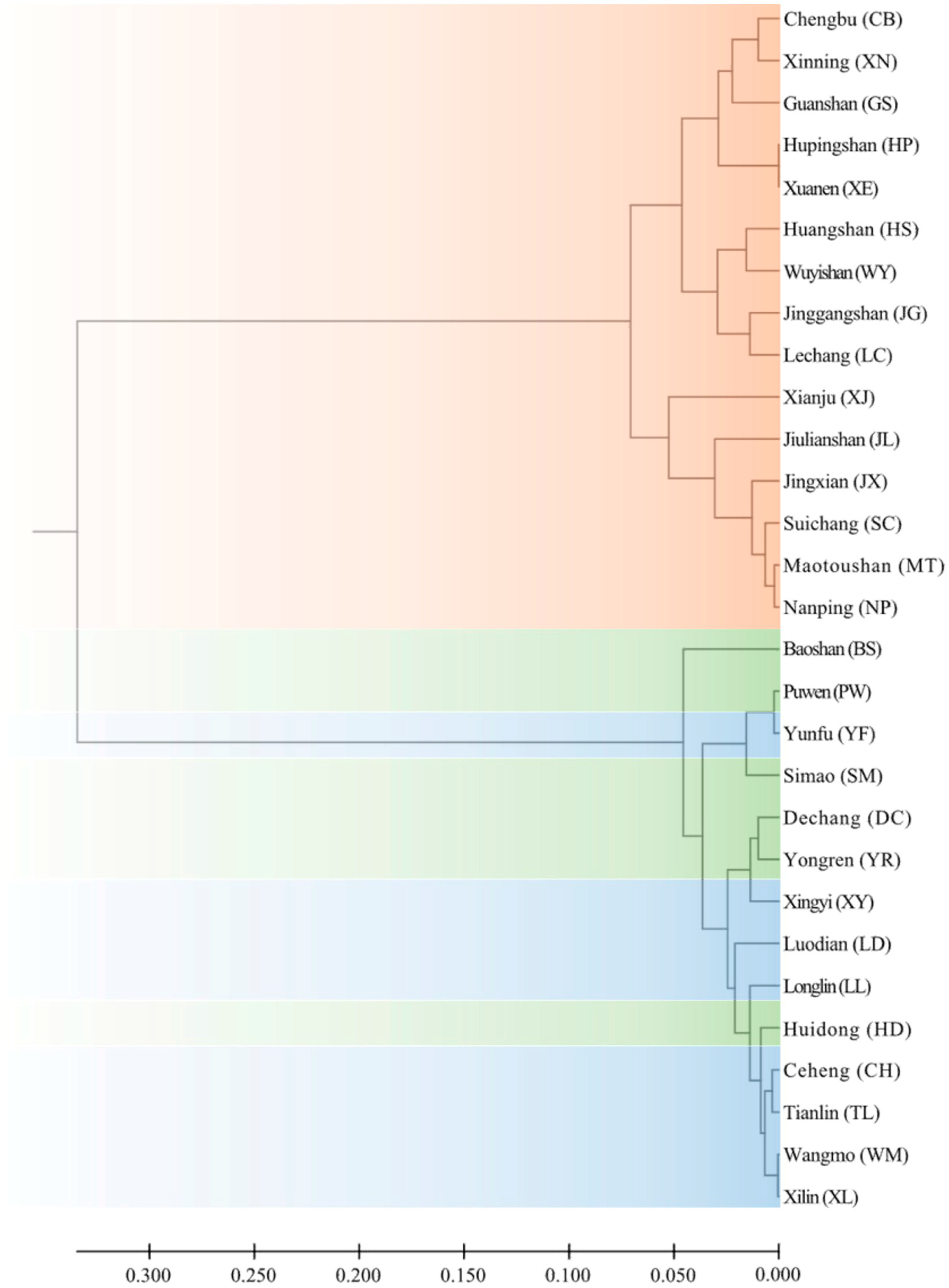
3.2. Population Genetic Structure
3.3. Relative Rate of Pollen to Seed Flow
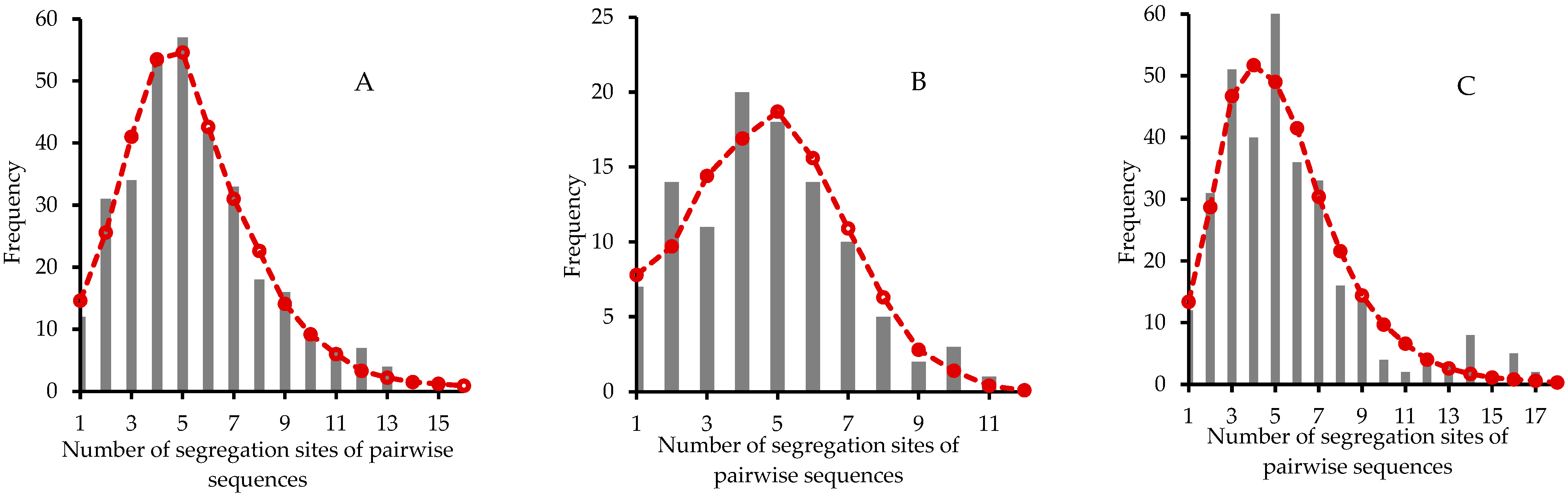
3.4. Population Demographic Analyses
4. Discussion
4.1. Genetic Diversity
4.2. Population Genetic Structure
4.3. Genetic Conservation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, S.K.; Li, H.; Chen, B.-Y. Meliaceae. In Flora of China 43; Science Press: Beijing, China, 1997; pp. 40–43. [Google Scholar]
- Styles, B.T. The flower biology of Meliaceae and its bearing on tree breeding. Silvae Genet. 1972, 21, 175–182. [Google Scholar]
- Edmonds, J.M. The potential value of Toona species (Meliaceae) as multipurpose and plantation trees in Southeast Asia. Commonw. For. Rev. 1993, 72, 181–186. [Google Scholar]
- Edmonds, J.M.; Staniforth, M. Toona sinensis (Meliaceae). Curtiss Bot. Mag. 1998, 15, 186–193. [Google Scholar] [CrossRef]
- Cheng, D.S.; Cui, T.L. Utilization value and cultivation techniques of Toona sureni. For. By-Prod. Spec. China 2010, 4, 39–40. [Google Scholar] [CrossRef]
- Fu, L.K.; Jin, J.M. China Plant Red Data Book: Rare and Endangered Plants; Science Press: Beijing, China, 1992; Volume 1. [Google Scholar]
- Liang, R.L.; Liao, R.Y.; Dai, J. Endangered causes and protection strategy of Toona ciliata. Guangxi For. Sci. 2011, 40, 201–203. [Google Scholar] [CrossRef]
- Li, P.; Que, Q.M.; Wu, L.Y.; Zhu, Q.; Chen, X.Y. Growth rhythms of Toona ciliata seedlings from different provenances. J. South China Agri. Univ. 2017, 38, 96–102. [Google Scholar] [CrossRef]
- Liao, D.; Wu, J.; Shu, Y.; Cheng, Y.; Chen, M.; Chen, J.; Liu, Q.; Wu, Z. Tissue culture of Toona ciliata. Agric. Sci. Technol. 2017, 18, 2185–2187, 2196. [Google Scholar] [CrossRef]
- Lan, J.H.; Feng, L.X. Study on cutting propagation technology of rare timber species Toona ciliata Roem. J. Guangxi Agric. 2022, 37, 32–36, 48. [Google Scholar] [CrossRef]
- Ennos, R.A.; French, G.C.; Hollingsworth, P.M. Conserving taxonomic complexity. Trends Ecol. Evol. 2005, 20, 164–168. [Google Scholar] [CrossRef]
- Li, P.; Zhan, X.; Que, Q.M.; Qu, W.T.; Liu, M.Q.; OuYang, K.X.; Li, J.C.; Deng, X.M.; Zhang, J.J.; Liao, B.Y.; et al. Genetic diversity and population structure of Toona ciliata Roem. based on sequence-related amplified polymorphism (SRAP) markers. Forests 2015, 6, 1094–1106. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Li, P.; Hui, W.K.; Deng, Y.W.; Gan, S.M.; Sun, Y.; Zhao, X.H.; Chen, X.Y.; Deng, X.M. Genetic diversity and population structure of Toona ciliata revealed by simple sequence repeat markers. Biotechnol. Biotechnol. Equip. 2019, 33, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Jiang, J.M.; Chen, Y.T. Genetic diversity of central and peripheral populations of Toona ciliata var. pubescens, an endangered tree species endemic to China. Genet. Mol. Res. 2014, 13, 4579–4590. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, X.X.; Ren, Y.; Li, P.; Chen, X.Y.; Hu, X.S. Mating system and population structure in the natural distribution of Toona ciliata (Meliaceae) in South China. Sci. Rep. 2020, 10, 16998. [Google Scholar] [CrossRef]
- Mullner, A.N.; Pennington, T.D.; Koecke, A.V.; Renner, S.S. Biogeography of Cedrela (Mellaceae, Sapindales) in central and south America. Am. J. Bot. 2010, 97, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Cavers, S.; Telford, A.; Arenal Cruz, F.; Perez Castaneda, A.J.; Valencia, R.; Navarro, C.; Buonamici, A.; Lowe, A.J.; Vendramin, G.G. Cryptic species and phylogeographical structure in the tree Cedrela odorata L. throughout the netotropics. J. Biogeogr. 2013, 40, 732–746. [Google Scholar] [CrossRef] [Green Version]
- Koecke, V.; Muellner-Riehl, A.N.; Pennington, T.D.; Schorr, G.; Schnitzler, J. Niche evolution through time and across continents: The story of neotropical Cedrela (Meliaceae). Am. J. Bot. 2013, 100, 1800–1810. [Google Scholar] [CrossRef]
- Koenen, E.J.; Clarkson, J.J.; Pennington, T.D.; Chatrou, L.W. Recently evolved diversity and convergent radiations of rainforest mahoganies (Meliaceae) shed new light on the origins of rainforest hyperdiversity. New Phytol. 2015, 207, 327–339. [Google Scholar] [CrossRef]
- Wang, X.; Xiao, Y.; He, Z.H.; Li, L.L.; Song, H.Y.; Zhang, J.J.; Cheng, X.; Chen, X.Y.; Li, P.; Hu, X.S. A chromosome-level genome assembly of Toona ciliata (Meliaceae). Genome Biol. Evol. 2022, 14, evac121. [Google Scholar] [CrossRef]
- Wang, X.; Xiao, Y.; He, Z.H.; Li, L.L.; Lv, Y.W.; Hu, X.S. Evolutionary divergence between Toona ciliata and Toona sinensis assayed with their whole genome sequences. Genes 2022, 13, 1799. [Google Scholar] [CrossRef]
- Hern, G.; Zhang, F.; Gao, Q.; Fu, P.C.; Xing, R.; Wang, J.; Liu, H.; Chen, S. Molecular phylogeography and intraspecific divergence of Spiraea alpina (Rosaceae) distributed in the Qinghai-Tibetan Plateau and adjacent regions inferred from nrDNA. Biochem. Syst. Ecol. 2014, 57, 278–286. [Google Scholar] [CrossRef]
- Sun, L.; Liu, G.; Lu, Y.; Zhang, B.; Zhang, G. Molecular data and ecological niche modelling reveal the phylogeographic pattern of the widespread shrub Tamarix chinensis Lour. (Tamaricaceae) in China. Kew Bull. 2020, 75, 41. [Google Scholar] [CrossRef]
- Freeland, J.R.; Petersen, S.D.; Kirk, H. Molecular Ecology; John Wiley & Sons, Ltd.: Publication, UK, 2011. [Google Scholar]
- Ennos, R.A. Estimating the relative rates of pollen and seed migration among plant populations. Heredity 1994, 72, 250–259. [Google Scholar] [CrossRef]
- Hu, X.S.; Ennos, R.A. Impacts of seed and pollen flow on population differentiation for plant genomes with three contrasting modes of inheritance. Genetics 1999, 152, 441–450. [Google Scholar] [CrossRef]
- Nota, K.; Klaminder, J.; Milesi, P.; Bindler, R.; Nobile, A.; van Steijn, T.; Bertilsson, S.; Svensson, B.; Hirota, S.K.; Matsuo, A.; et al. Norway spruce postglacial recolonization of Fennoscandia. Nat. Commun. 2022, 13, 1333. [Google Scholar] [CrossRef]
- Schulte, L.; Meucci, S.; Stoof-Leichsenring, K.R.; Heitkam, T.; Schmidt, N.; von Hippel, B.; Andreev, A.A.; Diekmann, B.; Biskaborn, B.K.; Wagner, B.; et al. Larix species range dynamics in Siberia since the Last Glacial captured from sedimentary ancient DNA. Commun. Biol. 2022, 5, 570. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, F.F.; Ma, S.M.; Wang, C.C.; Wei, B.; Zhang, Y.L. Phylogeography of Amygdalus mongolica in relation to Quaternary climatic aridification and oscillations in northwestern China. PeerJ 2022, 10, 13345. [Google Scholar] [CrossRef]
- Kirschner, P.; Perez, M.F.; Zaveska, E.; Sanmartin, I.; Marquer, L.; Schlick-Steiner, B.C.; Alvarez, N.; Steiner, F.M.; Schonswetter, P. Congruent evolutionary responses of European steppe biota to late Quaternary climate change. Nat. Commun. 2022, 13, 1921. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf material. Phytochemistry 1987, 19, 11–15. [Google Scholar]
- Demesure, B.; Sodzi, N.; Petit, R.J. A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol. Ecol. 1995, 4, 129–131. [Google Scholar] [CrossRef]
- Dumolin-Lapegue, S.; Pemonge, M.H.; Petit, R.J. An enlarged set of consensus primers for the study of organelle DNA in plants. Mol. Ecol. 1997, 6, 393–397. [Google Scholar] [CrossRef]
- Duminil, J.; Pemonge, M.H.; Petit, R.J. A set of 35 consensus primer pairs amplifying genes and introns of plant mitochondrial DNA. Mol. Ecol. Notes 2002, 2, 428–430. [Google Scholar] [CrossRef]
- Mishra, P.; Kumar, A.; Rodrigues, V.; Shukla, A.K.; Sundaresan, V. Feasibility of nuclear ribosomal region ITS1 over ITS2 in barcoding taxonomically challenging genera of subtribe Cassiinae (Fabaceae). PeerJ 2016, 4, e2638. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Watterson, G.A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975, 7, 188–193. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1660. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1951, 15, 323–354. [Google Scholar] [CrossRef]
- Pons, O.; Petitt, R.J. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 1996, 144, 1237–1245. [Google Scholar] [CrossRef]
- Grivet, D.; Petit, R.J. Chloroplast DNA phylogeography of the hornbeam in Europe: Evidence for a bottleneck at the outset of postglacial colonization. Conserv. Genet. 2002, 4, 47–56. [Google Scholar] [CrossRef]
- Meirmans, P.G. Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 2006, 60, 2399–2402. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef]
- Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 1997, 145, 1219–1228. [Google Scholar] [CrossRef]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 109–220. [Google Scholar]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the Genetics of Populations; University Chicago Press: Chicago, IL, USA, 1969; pp. 1191–1192. [Google Scholar]
- Hu, X.S.; Yeh, F.C.; Hu, Y.; Deng, L.T.; Ennos, R.A.; Chen, X.Y. High mutation rates explain low population genetic divergence at copy-number-variable loci in Homo sapiens. Sci. Rep. 2017, 7, 43178. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.; Walsh, B. Genetics and Analysis of Quantitative Traits; Sinauer Associates: Sunderland, UK, 1998; pp. 817–819. [Google Scholar]
- Goudet, J. FSTAT v-1.2: A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Yan, H.F.; Zhang, C.Y.; Wang, F.Y.; Hu, C.M.; Ge, X.J.; Hao, G. Population expanding with the phalanx model and lineages split by environmental heterogeneity: A case study of Primula obconica in subtropical China. PLoS ONE 2012, 7, e41315. [Google Scholar] [CrossRef]
- Miao, C.Y.; Yang, J.; Mao, R.L.; Li, Y. Phylogeography of Achyranthes bidentata (Amaranthaceae) in China’s warm-temperate zone inferred from chloroplast and nuclear DNA: Insights into population dynamics in response to climate change during the Pleistocene. Plant Mol. Biol. Rep. 2017, 35, 166–176. [Google Scholar] [CrossRef]
- Hernández-Rosales, H.S.; Castellanos-Morales, G.; Sánchez-de la Vega, G.; Aguirre-Planter, E.; Montes-Hernández, S.; Lira-Saade, R.; Eguiarte, L.E. Phylogeographic and population genetic analyses of Cucurbita moschata reveal divergence of two mitochondrial lineages linked to an elevational gradient. Am. J. Bot. 2020, 107, 510–525. [Google Scholar] [CrossRef]
- Muller, M.H.; Prosoeri, M.; Santoni, S.; Ronfort, J. Inferences from mitochondrial DNA patterns on the domestication history of alfalfa (Medicago sativa). Mol. Ecol. 2003, 12, 2187–2199. [Google Scholar] [CrossRef] [Green Version]
- Tomaru, N.; Takahashi, M.; Tsumura, Y.; Takahashi, M.; Ohba, K. Intraspecific variation and phylogeographic patterns of Fagus crenata (Fagaceae) mitochondrial DNA. Am. J. Bot. 1998, 85, 629–636. [Google Scholar] [CrossRef]
- Zhu, Q.; Liao, B.Y.; Li, P.; Li, J.C.; Deng, X.M.; Hu, X.S.; Chen, X.Y. Phylogeographic pattern suggests a major eastward dispersal in the distribution of Machilus pauhoi in South China. PLoS ONE 2017, 12, 0184456. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y. Phylogeographic Structure of Toona ciliata (Meliaceae) in China Inferred from cpDNA Markers and Ecological Niche Model Analyses. Master’s Thesis, South China Agricultural University, Guangzhou, China, 2019. [Google Scholar] [CrossRef]
- Tovar, C.; Carril, A.F.; Gutierrez, A.G.; Ahrends, A.; Fita, L.; Zaninell, P.; Flombaum, P.; Abarzúa, A.M.; Alarcón, D.; Aschero, V.; et al. Understanding climate change impacts on biome and plant distributions in the Andes: Challenges and opportunities. J. Biogeogr. 2022, 49, 1420–1442. [Google Scholar] [CrossRef]
- Zhang, C.H.; He, J.; Sun, Y.Y.; Li, K. Prediction of distributional change of Toona ciliata var. ciliata and application in regionalization of introduction based on MaxEnt. J. Yunnan Univ. 2018, 40, 164–173. [Google Scholar] [CrossRef]
- Zhang, C.H.; He, J.; Sun, Y.Y.; Li, K. Distributional change in suitable areas for T. ciliata var. pubescens based on MaxEnt. For. Res. 2018, 31, 120–126. [Google Scholar] [CrossRef]
- Hu, X.S.; Wu, R.L.; Han, Y.F. An approach to sustainable management of population genetic resources of trees: (II) Management analyses of plantation and natural population of some autochthonous tree species in China. For. Res. 2001, 14, 1–7. [Google Scholar]
- Salmela, M.J.; Cavers, S.; Wachowiak, W.; Cottrell, J.E.; Iason, G.R.; Ennos, R.A. Understanding the evolution of native pinewoods in Scotland will benefit their future management and conservation. Forestry 2010, 83, 535–545. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Code | Sample Size | Longitude (E) | Latitude (N) | Elevation (m) |
|---|---|---|---|---|---|
| Huangshan, Anhui | HS | 6 | 118.08 | 30.16 | 499 |
| Jingxian, Anhui | JX | 15 | 118.24 | 30.41 | 366 |
| Nanping, Fujian | NP | 10 | 118.10 | 26.38 | 800 |
| Lechang, Guangdong | LC | 15 | 113.20 | 25.07 | 359 |
| Yunfu, Guangdong | YF | 18 | 111.34 | 22.46 | 340 |
| Longlin, Guangxi | LL | 10 | 105.20 | 24.46 | 624 |
| Tianlin, Guangxi | TL | 26 | 106.13 | 24.17 | 792 |
| Xilin, Guangxi | XL | 27 | 105.05 | 24.29 | 899 |
| Luodian, Guizhou | LD | 14 | 106.44 | 25.25 | 814 |
| Ceheng, Guizhou | CH | 22 | 105.48 | 24.59 | 730 |
| Wangmo, Guizhou | WM | 30 | 106.05 | 25.10 | 500 |
| Xingyi, Guizhou | XY | 15 | 104.54 | 25.06 | 1160 |
| Xuanen, Hubei | XE | 9 | 109.28 | 30.17 | 632 |
| Chengbu, Hunan | CB | 25 | 111.28 | 27.14 | 737 |
| Hupingshan, Hunan | HP | 3 | 111.41 | 29.01 | 436 |
| Xinning, Hunan | XN | 30 | 109.44 | 28.18 | 650 |
| Maotoushan, Jiangxi | MT | 14 | 117.03 | 27.41 | 1000 |
| Wuyishan, Fujian | WY | 9 | 117.42 | 28.18 | 1200 |
| Guanshan, Jiangxi | GS | 14 | 115.26 | 27.19 | 337 |
| Jinggangshan, Jiangxi | JG | 17 | 114.17 | 26.44 | 400 |
| Jiulianshan, Jiangxi | JL | 18 | 114.47 | 24.54 | 610 |
| Dechang, Sichuan | DC | 29 | 102.32 | 26.40 | 1325 |
| Huidong, Sichuan | HD | 30 | 102.09 | 27.23 | 1862 |
| Baoshan, Yunnan | BS | 11 | 99.06 | 25.04 | 1513 |
| Yongren, Yunnan | YR | 29 | 101.32 | 25.01 | 1539 |
| Simao, Yunnan | SM | 16 | 100.58 | 22.46 | 1317 |
| Puwen, Yunnan | PW | 19 | 101.04 | 22.23 | 890 |
| Xianju, Zhejiang | XJ | 7 | 119.92 | 28.45 | 600 |
| Suichang, Zhejiang | SC | 12 | 119.25 | 28.59 | 510 |
| Population | mtDNA Marker | Haploid nrDNA ITS | Tajima’s D (p-Value) | Fu’s F (p-Value) | ||
|---|---|---|---|---|---|---|
| h | π | π | θ | |||
| HS | 0.500 | 0.00108 | 0.02700 | 0.036 | −0.73 (0.29) | 2.07 (0.76) |
| JX | 0.124 | 0.00032 | 0.00888 | 0.018 | −1.79 (0.03) | −0.75 (0.35) |
| NP | 0.500 | 0.00045 | 0.00413 | 0.006 | −0.76 (0.24) | −1.54 (0.14) |
| LC | 0.240 | 0.00065 | 0.01512 | 0.019 | −0.88 (0.18) | −1.11 (0.27) |
| YF | 0.401 | 0.00171 | 0.00906 | 0.014 | −1.50 (0.04) | −1.00 (0.31) |
| LL | 0.000 | 0.0000 | 0.00528 | 0.008 | −0.35 (0.41) | −0.88 (0.28) |
| TL | 0.074 | 0.00019 | 0.00609 | 0.009 | −0.93 (0.21) | −16.71 (0.00) |
| XL | 0.000 | 0.0000 | 0.00542 | 0.013 | −1.90 (0.02) | −9.32 (0.00) |
| LD | 0.000 | 0.0000 | 0.00481 | 0.008 | −1.58 (0.04) | −4.70 (0.00) |
| CH | 0.000 | 0.0000 | 0.00691 | 0.011 | −0.98 (0.19) | −9.27 (0.00) |
| WM | 0.180 | 0.00015 | 0.00614 | 0.014 | −1.88 (0.01) | −10.88 (0.00) |
| XY | 0.000 | 0.0000 | 0.01136 | 0.019 | −1.36 (0.08) | −8.81 (0.00) |
| XE | 0.346 | 0.00031 | 0.00186 | 0.003 | −0.80 (0.24) | −0.40 (0.38) |
| CB | 0.403 | 0.00034 | 0.00617 | 0.010 | −1.15 (0.14) | −2.22 (0.17) |
| HP | 0.000 | 0.0000 | 0.00220 | 0.003 | NA | NA |
| XN | 0.000 | 0.0000 | 0.00266 | 0.005 | −1.16 (0.12) | 1.31 (0.79) |
| MT | 0.000 | 0.0000 | 0.00246 | 0.003 | −0.20 (0.62) | 1.31 (0.77) |
| WY | 0.370 | 0.00148 | 0.02526 | 0.032 | −0.65 (0.29) | 1.62 (0.73) |
| GS | 0.459 | 0.00108 | 0.00360 | 0.006 | −1.24 (0.10) | −1.41 (0.18) |
| JG | 0.381 | 0.00173 | 0.01811 | 0.026 | −1.11 (0.11) | −0.09 (0.46) |
| JL | 0.444 | 0.00038 | 0.00441 | 0.012 | −1.74 (0.03) | −1.73 (0.18) |
| DC | 0.186 | 0.00015 | 0.00786 | 0.014 | −1.27 (0.10) | −6.69 (0.01) |
| HD | 0.000 | 0.0000 | 0.00515 | 0.008 | −0.92 (0.20) | −8.37 (0.00) |
| BS | 0.000 | 0.0000 | 0.03222 | 0.038 | −0.72 (0.22) | 2.12 (0.83) |
| YR | 0.000 | 0.0000 | 0.00590 | 0.009 | −0.75 (0.26) | −7.17 (0.00) |
| SM | 0.508 | 0.00077 | 0.00515 | 0.008 | −1.52 (0.06) | −6.90 (0.00) |
| PW | 0.388 | 0.00033 | 0.00527 | 0.007 | −0.75 (0.25) | −0.59 (0.39) |
| XJ | 0.000 | 0.0000 | 0.00110 | 0.002 | −1.61 (0.04) | −0.13 (0.44) |
| SC | 0.000 | 0.0000 | 0.00330 | 0.005 | −0.18 (0.46) | −0.73 (0.34) |
| Marker | Source of Variation | d.f. | Sum of Square | Variance Component | Percentage of Variance (%) | Φst | p-Value |
|---|---|---|---|---|---|---|---|
| cox1−nad1 | Among populations | 28 | 1599.283 | 3.3129 | 88.58 | 0.8858 | 0.00 |
| Within populations | 471 | 201.153 | 0.4271 | 11.42 | |||
| Total | 499 | 1800.436 | 3.7400 | ||||
| 26S−rRNA−tRNA−Leu | Among populations | 28 | 2527.316 | 5.2370 | 89.01 | 0.8901 | 0.00 |
| Within populations | 471 | 304.612 | 0.6467 | 10.99 | |||
| Total | 499 | 2831.928 | 5.8837 | ||||
| cox1−nad1 and 26S−rRNA−tRNA−Leu | Among populations | 28 | 4126.600 | 8.5499 | 88.84 | 0.8884 | 0.00 |
| Within populations | 471 | 505.764 | 1.0738 | 11.16 | |||
| Total | 499 | 4632.364 | 9.6237 | ||||
| ITS | Among populations | 28 | 3192.408 | 6.9508 | 71.43 | 0.7143 | 0.00 |
| Within populations | 438 | 1217.771 | 2.7803 | 28.57 | |||
| Total | 466 | 4410.180 | 9.7311 |
| Western Region | Central Region | Western and Central Region | Eastern Region | Global Region | |
|---|---|---|---|---|---|
| Fst(n) | 0.1362 | 0.0506 | 0.1068 | 0.1810 | 0.6994 |
| Sd(Fst(n)) | 0.0016 | 0.0038 | 0.0003 | 0.0247 | 0.0079 |
| Fst(m) | 0.8405 | 0.0262 | 0.8566 | 0.5360 | 0.8870 |
| Sd(Fst(m)) | 0.0600 | 0.0011 | 0.0022 | 0.1267 | 0.0077 |
| mP/mS | 31.4134 | -1.4950 | 47.9628 | 3.2270 | 1.3741 |
| Sd(mP/mS) | 13.2330 | 0.0454 | 0.8091 | 2.5527 | 0.2925 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Zhang, X.-X.; Hu, Y.; Wang, X.; Li, P.; He, Z.-H.; Lv, Y.-W.; Chen, X.-Y.; Hu, X.-S. Phylogeography of Toona ciliata (Meliaceae) Complex in China Inferred from Cytonuclear Markers. Genes 2023, 14, 116. https://doi.org/10.3390/genes14010116
Xiao Y, Zhang X-X, Hu Y, Wang X, Li P, He Z-H, Lv Y-W, Chen X-Y, Hu X-S. Phylogeography of Toona ciliata (Meliaceae) Complex in China Inferred from Cytonuclear Markers. Genes. 2023; 14(1):116. https://doi.org/10.3390/genes14010116
Chicago/Turabian StyleXiao, Yu, Xin-Xin Zhang, Ying Hu, Xi Wang, Pei Li, Zi-Han He, Yan-Wen Lv, Xiao-Yang Chen, and Xin-Sheng Hu. 2023. "Phylogeography of Toona ciliata (Meliaceae) Complex in China Inferred from Cytonuclear Markers" Genes 14, no. 1: 116. https://doi.org/10.3390/genes14010116
APA StyleXiao, Y., Zhang, X.-X., Hu, Y., Wang, X., Li, P., He, Z.-H., Lv, Y.-W., Chen, X.-Y., & Hu, X.-S. (2023). Phylogeography of Toona ciliata (Meliaceae) Complex in China Inferred from Cytonuclear Markers. Genes, 14(1), 116. https://doi.org/10.3390/genes14010116