
An Aggrephagy-Related LncRNA Signature for the Prognosis of Pancreatic Adenocarcinoma
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source and Processing
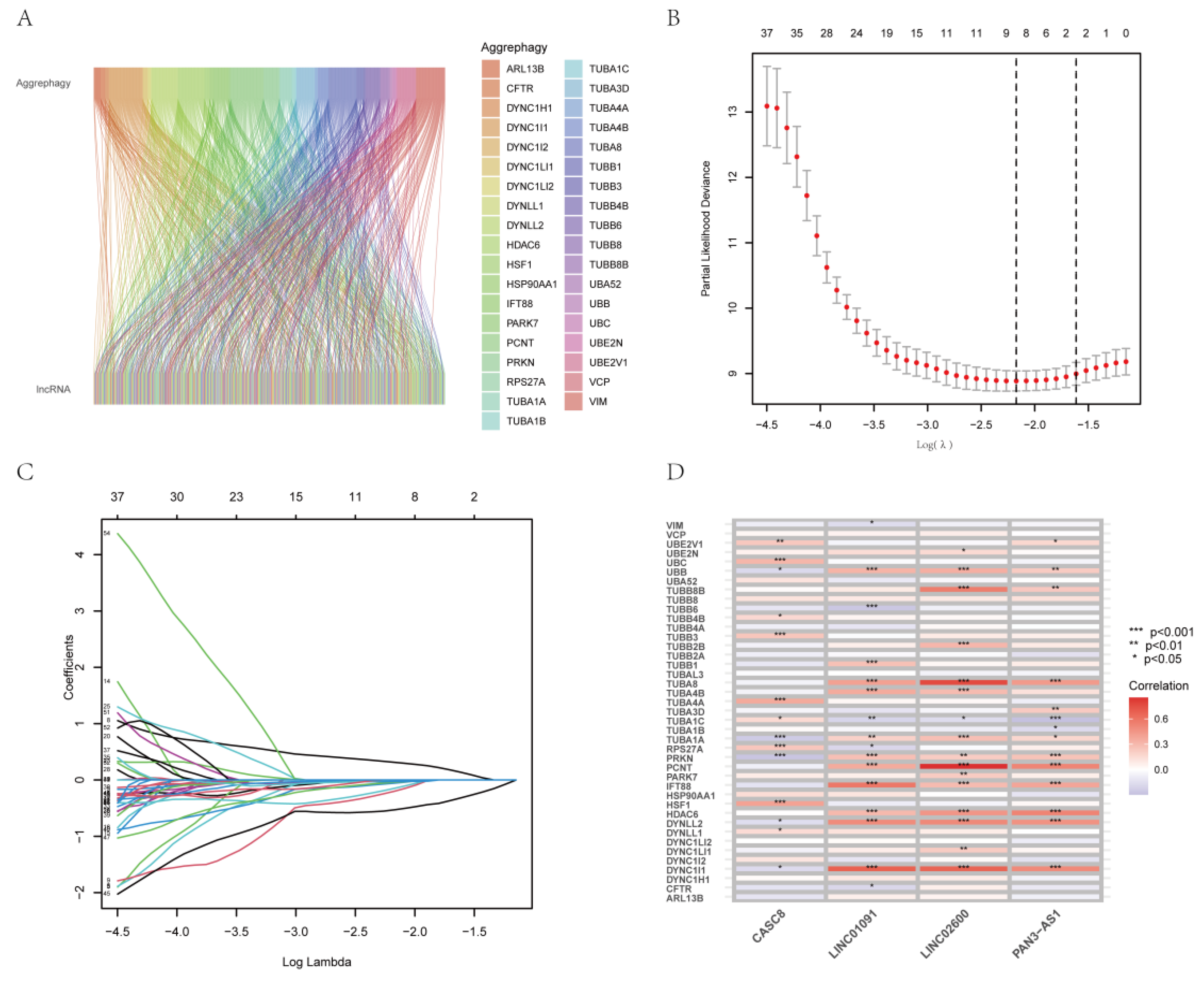
2.2. Screening of AGGLncRNAs
2.3. Development and Verification of an AGGLncRNA Prognostic Model
2.4. Construction of the Nomogram
2.5. Analysis of Functional Enrichment
2.6. The Risk Signature’s Immunity Analysis
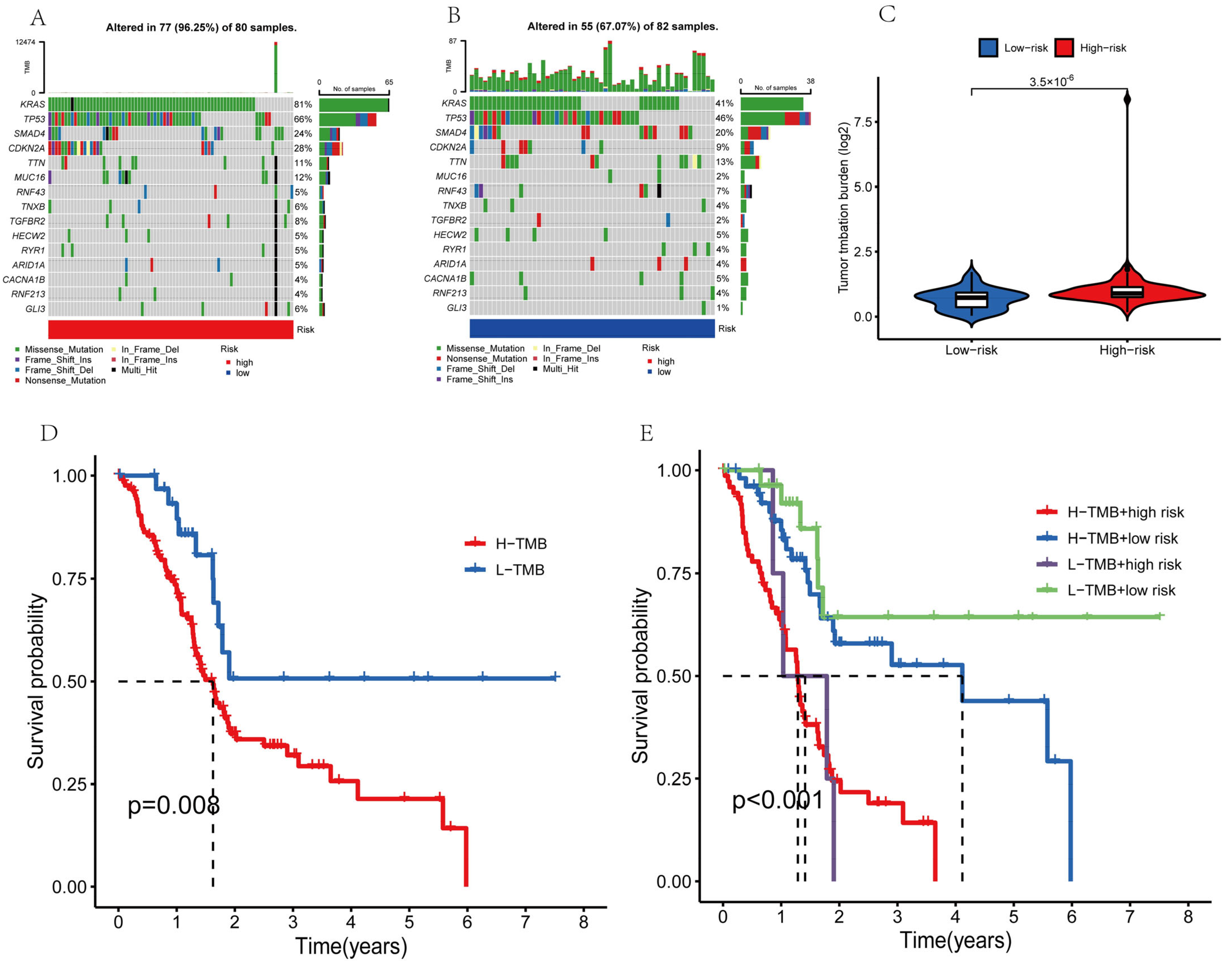
2.7. Analysis of Tumor Mutations
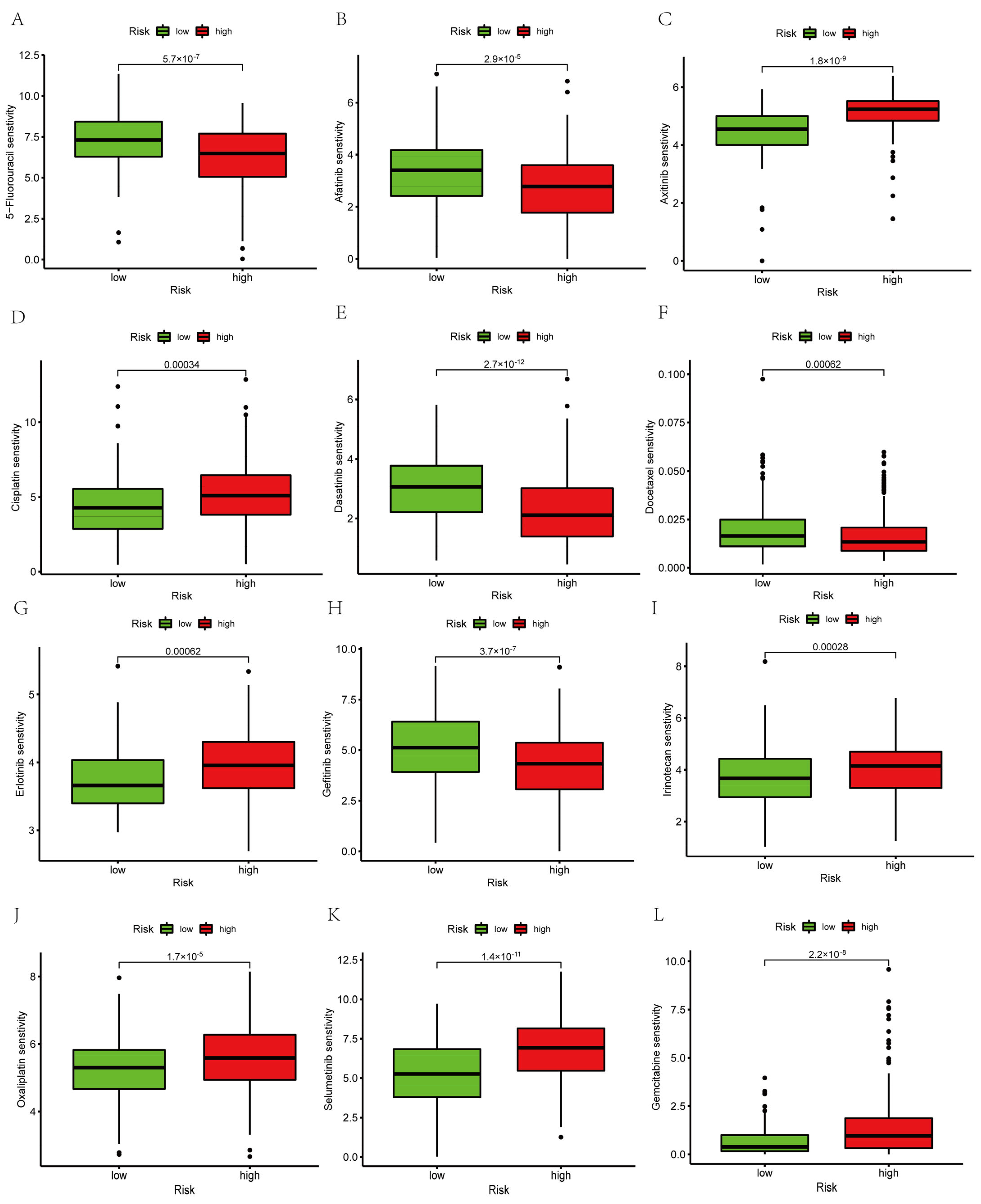
2.8. Prediction of Targeted Drug Sensitivity and Immunotherapy Response
2.9. Statistical Analysis
3. Results
3.1. Acquisition of Differentially Expressed AGGLncRNAs in PAAD
3.2. Construction of a Prognostic Characteristic Risk Score Model for AGGLncRNAs
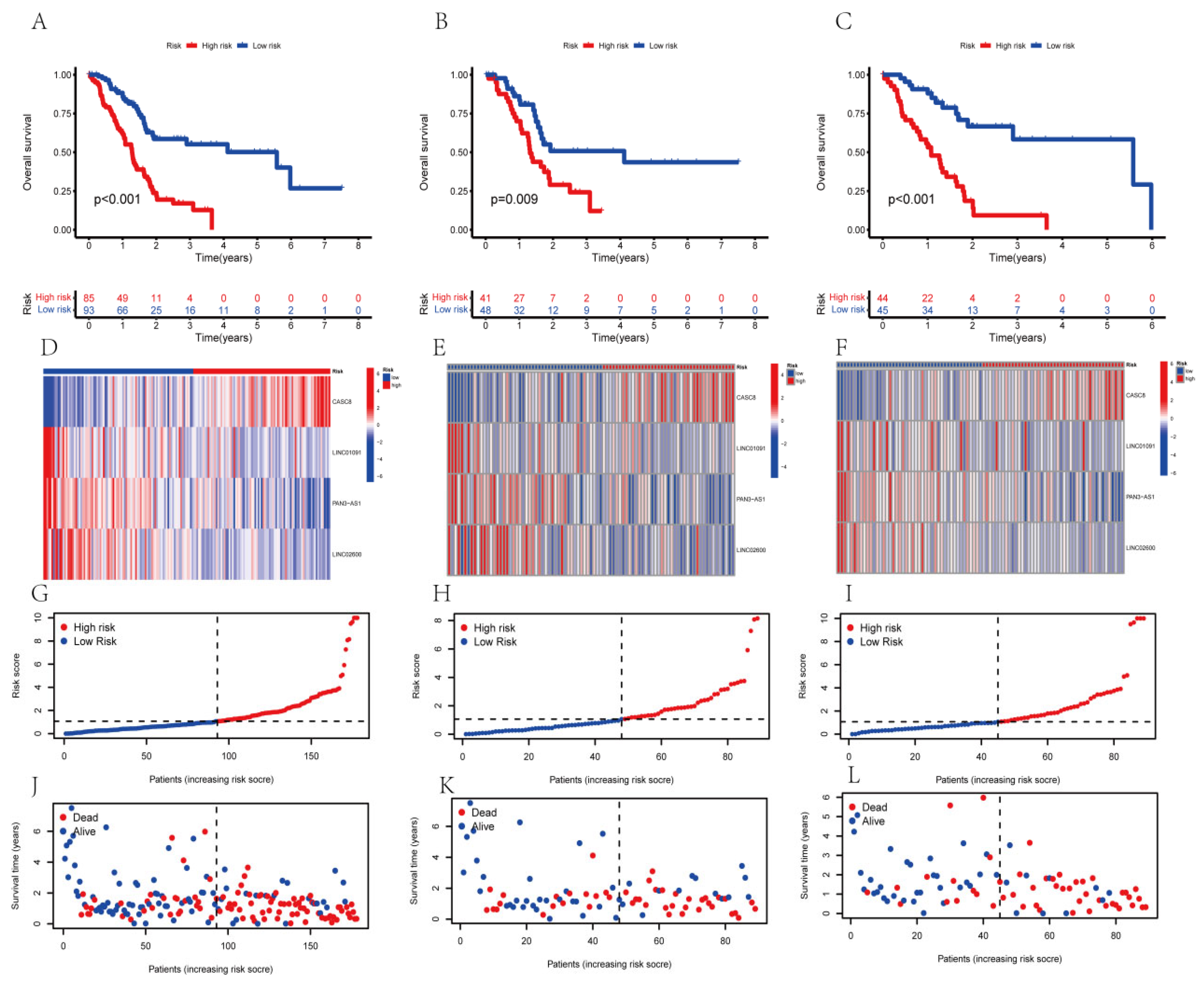
3.3. The Survival Analysis of the AGGLncRNAs Signature
3.4. Validation of the Ability of the AGGLncRNA Signature to Predict Prognosis
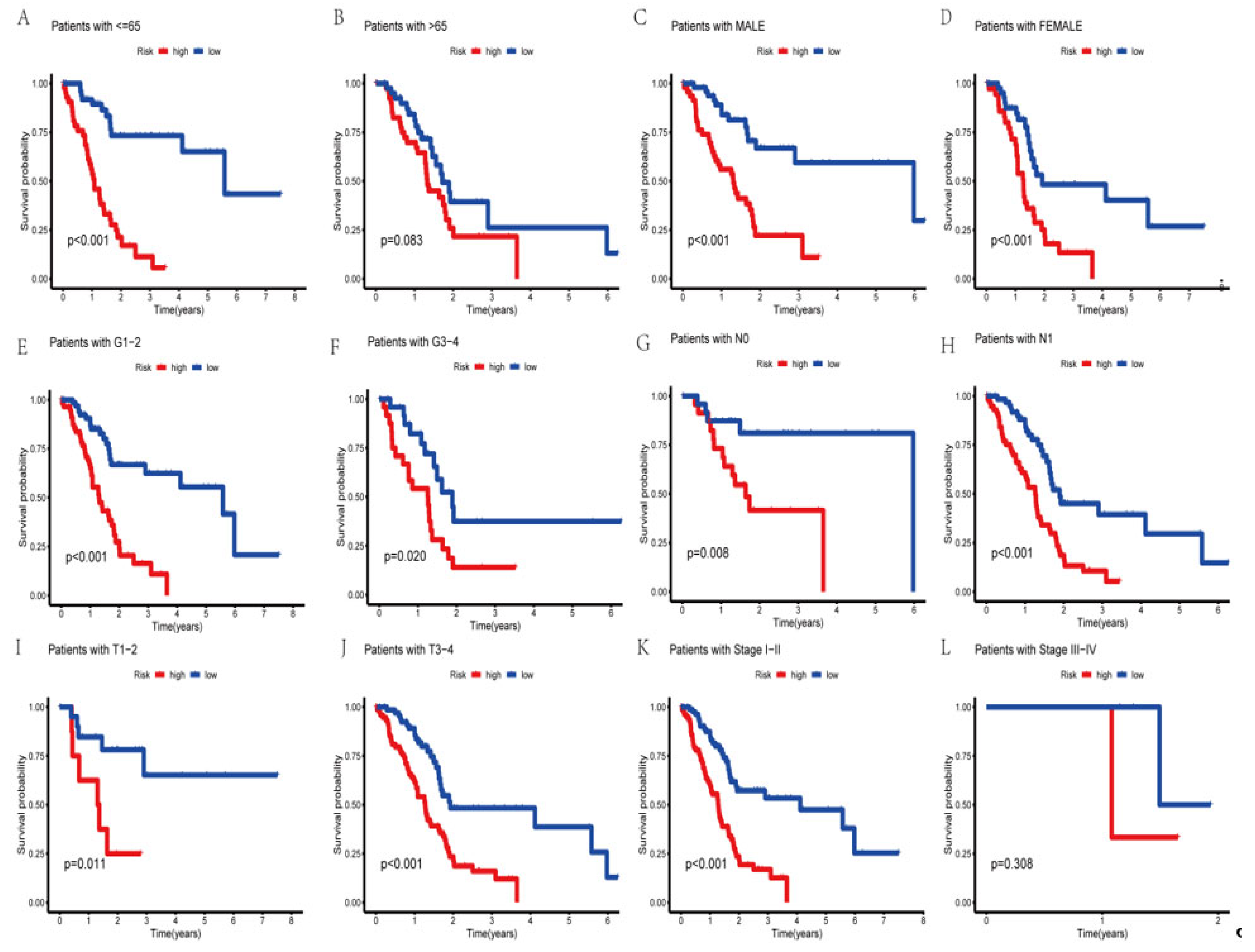
3.5. Survival Analysis of the Clinical Subgroups
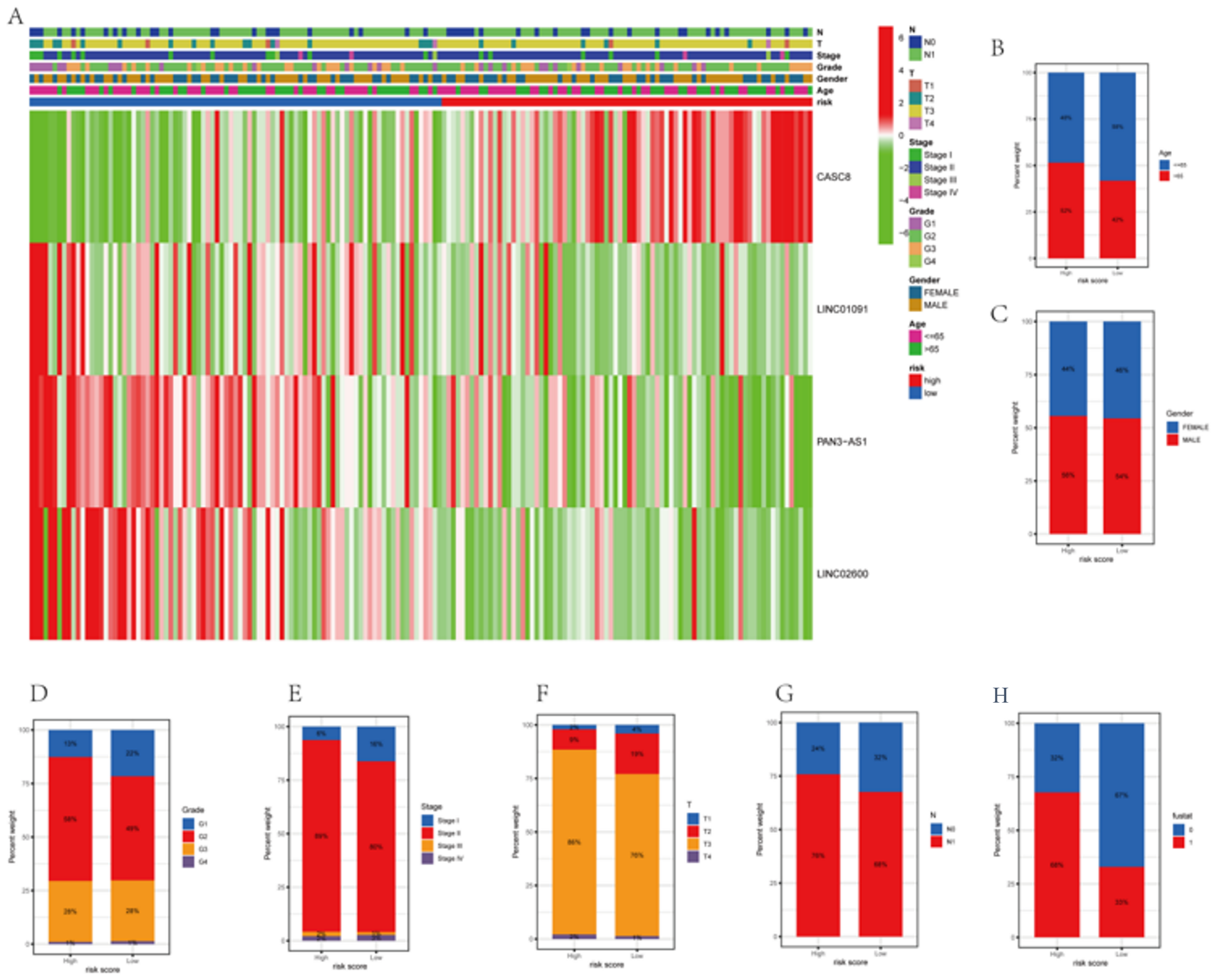
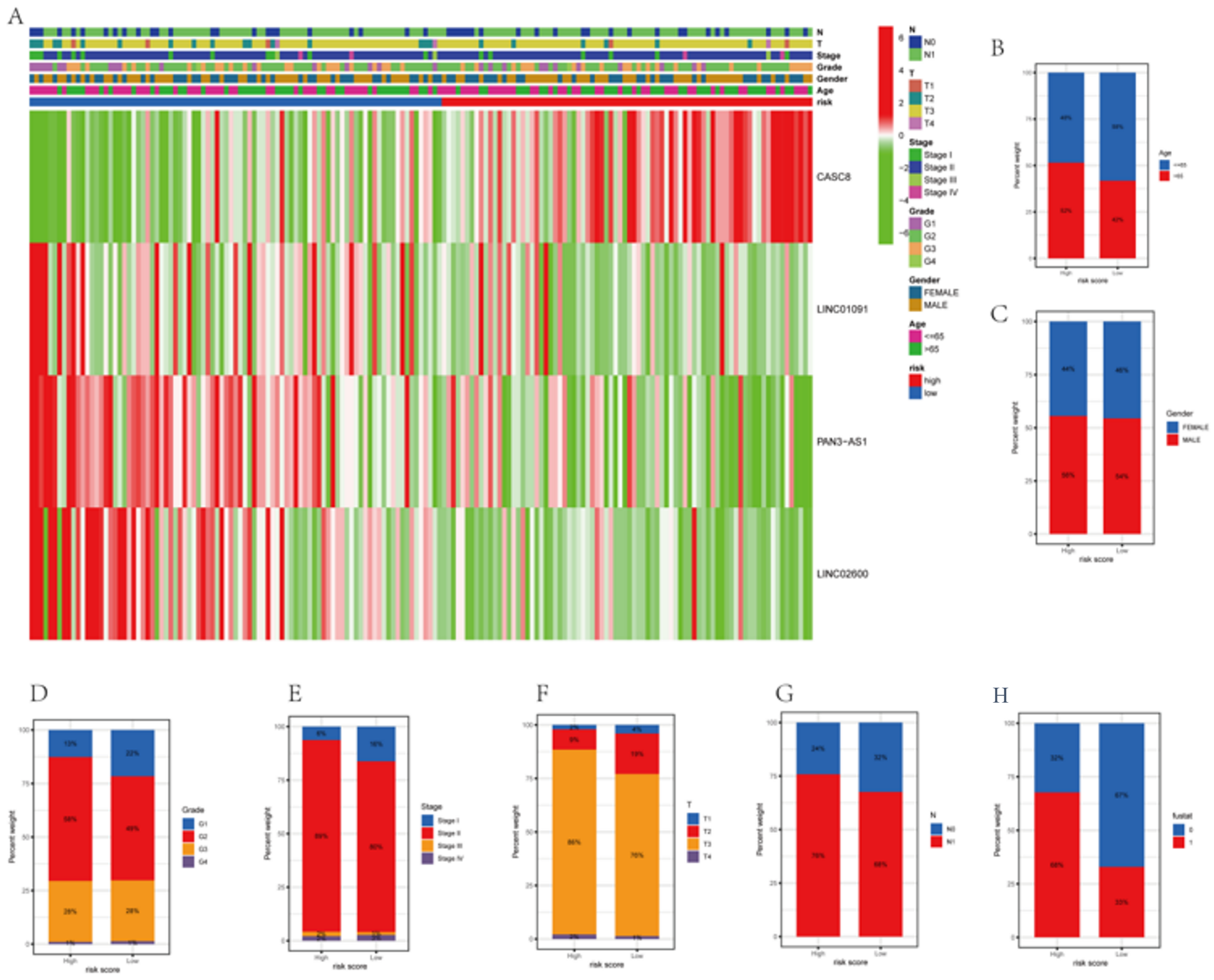
3.6. Correlations between Clinicopathological Characteristics and Risk Scores
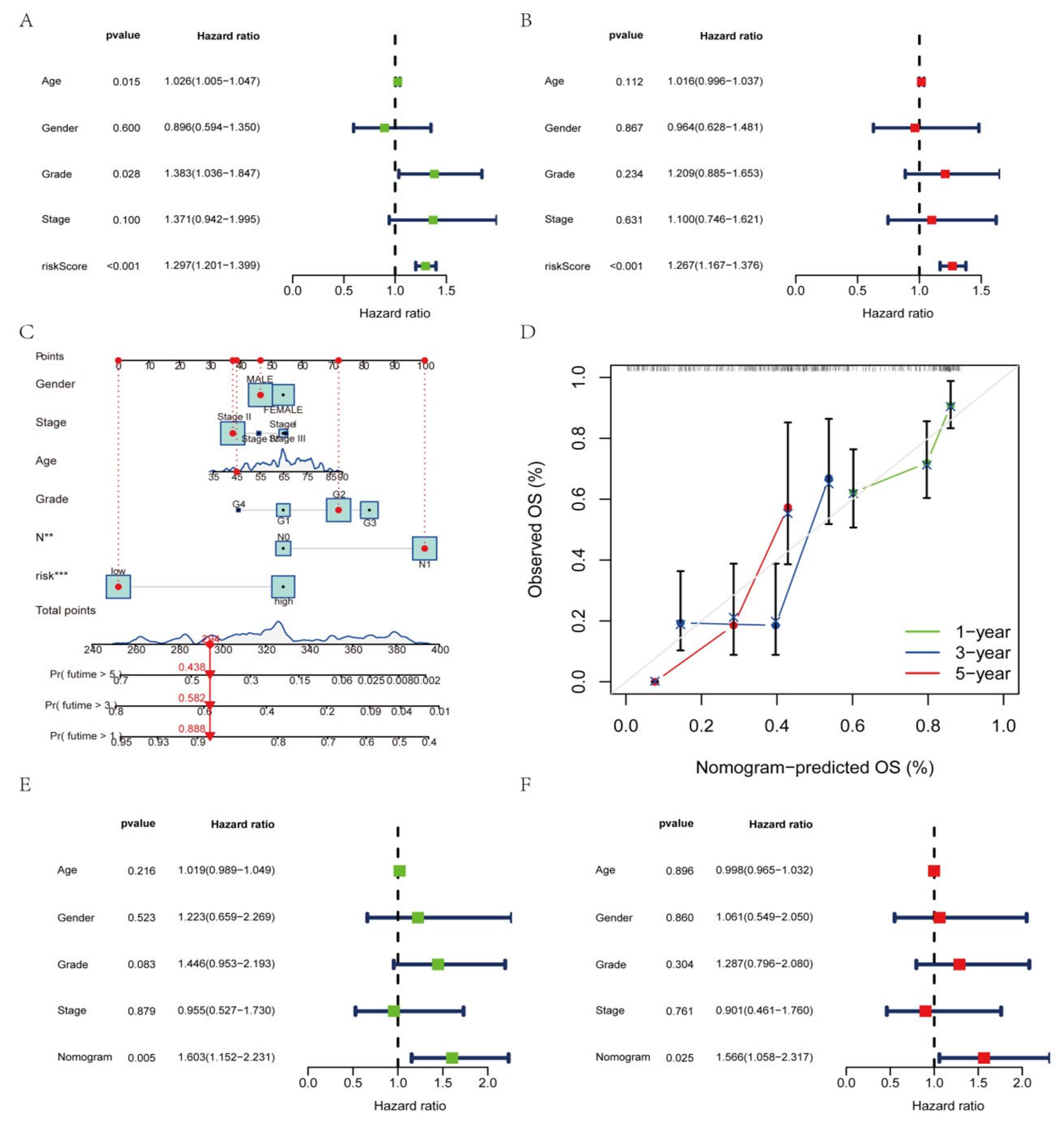
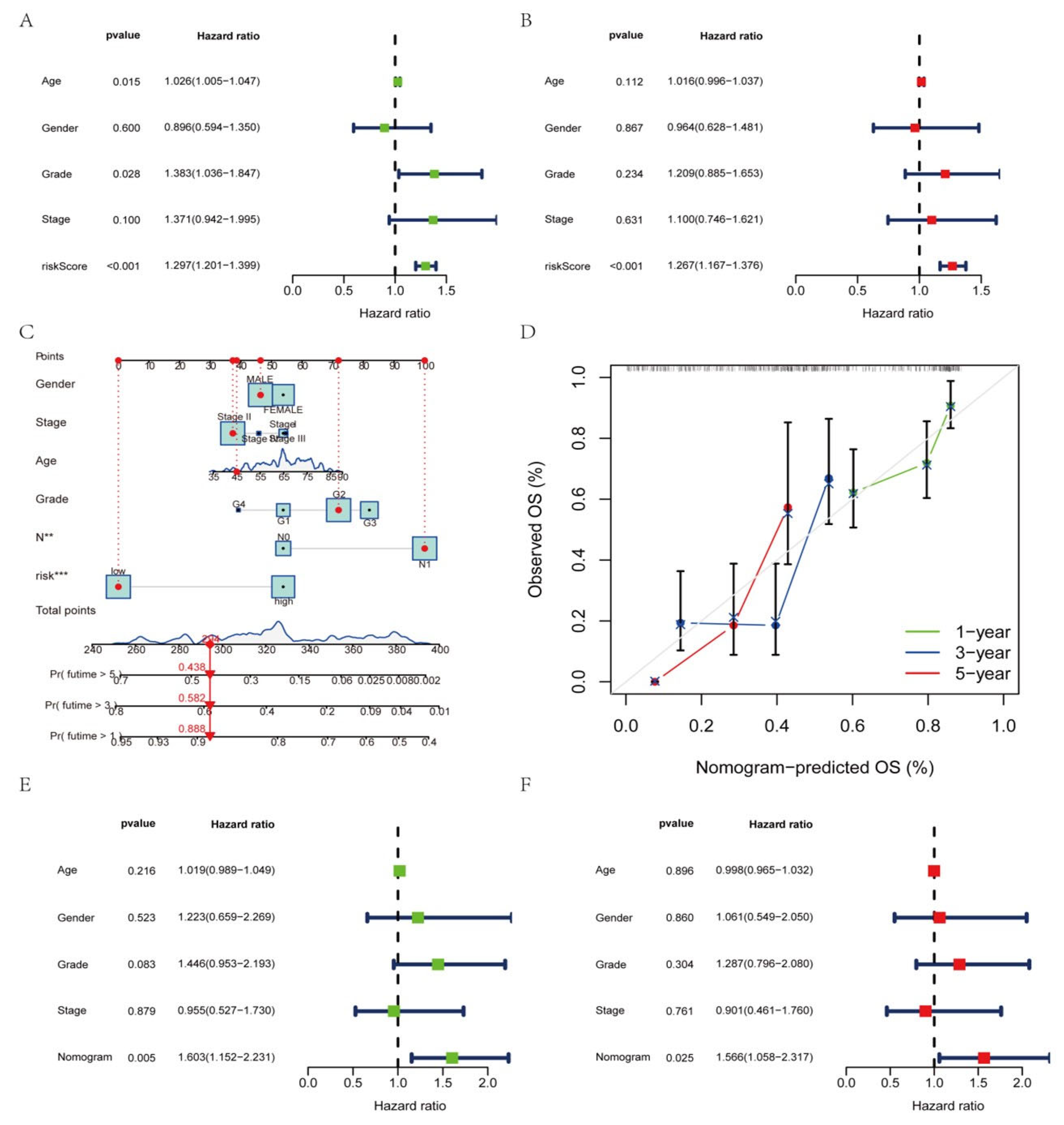
3.7. Construction of a Nomogram Based on Clinical Features
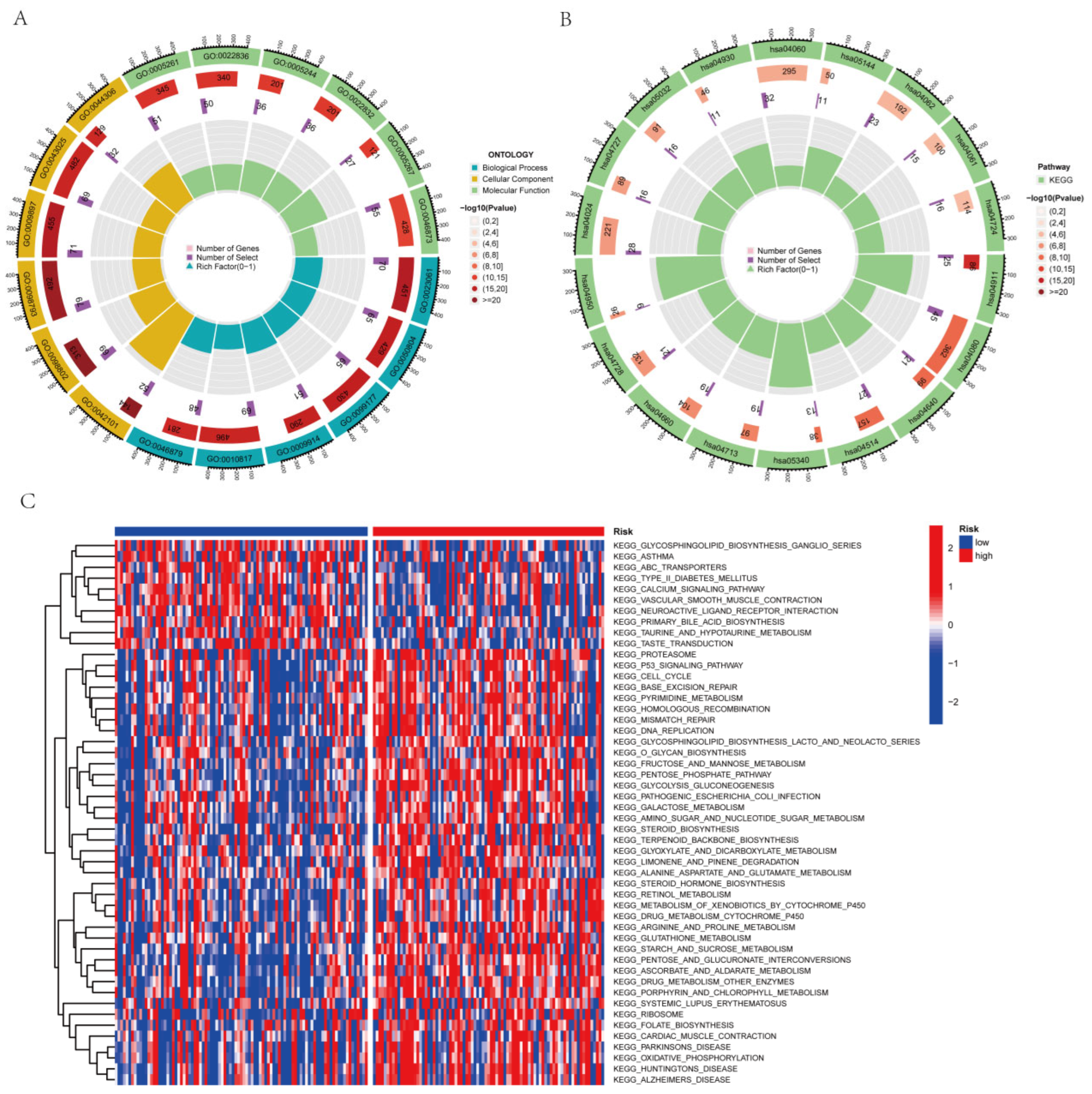
3.8. Assessment of Functional Enrichment
3.9. Predicting Immune Cell Infiltration and Response to Immunotherapy Based on the AGGLncRNA Signature
3.10. Evaluation of Tumor Mutations
3.11. Prediction of Targeted Drug Sensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Salmeron, C.; Wiley, S.Z.; Insel, P.A. GPCRs in pancreatic adenocarcinoma: Contributors to tumour biology and novel therapeutic targets. Br. J. Pharmacol. 2020, 177, 2434–2455. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.-S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromisch, C.; Qadan, M.; Machado, M.A.; Liu, K.; Colson, Y.; Grinstaff, M.W. Pancreatic Adenocarcinoma: Unconventional Approaches for an Unconventional Disease. Cancer Res 2020, 80, 3179–3192. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Ho, H.C.; Su, Y.C.; Yu, C.H.; Yang, C.C. Modified Tumor Classification With Inclusion of Tumor Characteristics Improves Discrimination and Prediction Accuracy in Oral and Hypopharyngeal Cancer Patients Who Underwent Surgery. Medicine 2015, 94, e1114. [Google Scholar] [CrossRef]
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.-L.; Malka, D.; Ducreux, M.; Conroy, T. An update on treatment options for pancreatic adenocarcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Gong, Q.; Dong, Q.; Zhong, B.; Zhang, T.; Cao, D.; Zhang, Y.; Ma, D.; Cai, X.; Li, Z. Clinicopathological features, prognostic significance, and associated tumor cell functions of family with sequence similarity 111 member B in pancreatic adenocarcinoma. J. Clin. Lab. Anal. 2022, 36, e24784. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef]
- Muscolino, E.; Schmitz, R.; Loroch, S.; Caragliano, E.; Schneider, C.; Rizzato, M.; Brune, W. Herpesviruses induce aggregation and selective autophagy of host signalling proteins NEMO and RIPK1 as an immune-evasion mechanism. Nat. Microbiol. 2020, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Shima, T.; Tsuda, S.; Aoki, A.; Kawaguchi, M.; Furuta, A.; Yasuda, I.; Yoneda, S.; Yamaki-Ushijima, A.; Cheng, S.-B.; et al. Aggrephagy Deficiency in the Placenta: A New Pathogenesis of Preeclampsia. Int. J. Mol. Sci. 2021, 22, 2432. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.B.; Sharma, S. Preeclampsia and health risks later in life: An immunological link. Semin. Immunopathol. 2016, 38, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science (N. Y., NY) 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, X.; Kang, R.; Zeh, H.; Klionsky, D.J.; Tang, D. Regulation and function of autophagy in pancreatic cancer. Autophagy 2021, 17, 3275–3296. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef]
- Jendrzejewski, J.; He, H.; Radomska, H.S.; Li, W.; Tomsic, J.; Liyanarachchi, S.; Davuluri, R.V.; Nagy, R.; de la Chapelle, A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc. Natl. Acad. Sci. USA 2012, 109, 8646–8651. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Fullwood, M.J. Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef]
- Yan, R.L.; Luan, C.L.; Liao, C.C.; Liu, L.H.; Chen, F.Y.; Chen, H.Y.; Chen, R.H. Long noncoding RNA BCRP3 stimulates VPS34 and autophagy activities to promote protein homeostasis and cell survival. J. Biomed. Sci. 2022, 29, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, Y.; Liu, X.; Long, Y.; Chen, J. LncRNA-Regulated Autophagy and its Potential Role in Drug-Induced Liver Injury. Ann. Hepatol. 2018, 17, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Zhang, Y.H.; Li, R.B.; Zhou, L.Y.; An, T.; Zhang, R.C.; Zhai, M.; Huang, Y.; Yan, K.-W.; Dong, Y.-H.; et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat. Commun. 2018, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Dienstmann, R.; Villacampa, G.; Sveen, A.; Mason, M.J.; Niedzwiecki, D.; Nesbakken, A.; Guinney, J. Relative contribution of clinicopathological variables, genomic markers, transcriptomic subtyping and microenvironment features for outcome prediction in stage II/III colorectal cancer. Ann. Oncol. 2019, 30, 1622–1629. [Google Scholar] [CrossRef] [Green Version]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017, 6, e26476. [Google Scholar] [CrossRef]
- Tamminga, M.; Hiltermann TJ, N.; Schuuring, E.; Timens, W.; Fehrmann, R.S.; Groen, H.J. Immune microenvironment composition in non-small cell lung cancer and its association with survival. Clin. Transl. Immunol. 2020, 9, e1142. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Zheng, S. Successful Immunotherapy for Pancreatic Cancer in a Patient With TSC2 and SMAD4 Mutations: A Case Report. Front. Immunol. 2021, 12, 785400. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Li, X.; Shi, Y.; Lu, Y.; Qiu, P.; Deng, Z.; Yao, W.; Wang, J. A Comprehensive Analysis of Pyroptosis-Related lncRNAs Signature Associated With Prognosis and Tumor Immune Microenvironment of Pancreatic Adenocarcinoma. Front. Genet. 2022, 13, 899496. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Xing, C.; Ding, C.; Zhang, H.; Chen, L.; You, L.; Dai, M.; Zhao, Y. Tumor microenvironment in chemoresistance, metastasis and immunotherapy of pancreatic cancer. Am. J. Cancer Res. 2020, 10, 1937–1953. [Google Scholar] [PubMed]
- Chi, H.; Peng, G.; Wang, R.; Yang, F.; Xie, X.; Zhang, J.; Tian, G. Cuprotosis Programmed-Cell-Death-Related lncRNA Signature Predicts Prognosis and Immune Landscape in PAAD Patients. Cells 2022, 11, 3436. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Reid, M.D.; Saka, B.; Balci, S.; Goldblum, A.S.; Adsay, N.V. Molecular genetics of pancreatic neoplasms and their morphologic correlates: An update on recent advances and potential diagnostic applications. Am. J. Clin. Pathol. 2014, 141, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Verbeke, C. Morphological heterogeneity in ductal adenocarcinoma of the pancreas—Does it matter? Pancreatology 2016, 16, 295–301. [Google Scholar] [CrossRef]
- Hong, S.M.; Park, J.Y.; Hruban, R.H.; Goggins, M. Molecular signatures of pancreatic cancer. Arch. Pathol. Lab. Med. 2011, 135, 716–727. [Google Scholar] [CrossRef]
- Canto, M.I.; Harinck, F.; Hruban, R.H.; Offerhaus, G.J.; Poley, J.W.; Kamel, I.; Nio, Y.; Schulick, R.; Bassi, C.; Kluijt, I.; et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013, 62, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Choi, K.S. Autophagy and cancer. Exp. Mol. Med. 2012, 44, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Taniue, K.; Akimitsu, N. The Functions and Unique Features of LncRNAs in Cancer Development and Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 632. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Huo, H.; You, Z.; Lu, R.; Yao, T.; Huang, J. Identification of cuproptosis-associated IncRNAs signature and establishment of a novel nomogram for prognosis of stomach adenocarcinoma. Front. Genet. 2022, 13, 982888. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhang, H.; Yang, D.; Min, Q.; Wang, Y.; Zhang, W.; Zhan, Q. The m6A-induced lncRNA CASC8 promotes proliferation and chemoresistance via upregulation of hnRNPL in esophageal squamous cell carcinoma. Int. J. Biol. Sci. 2022, 18, 4824–4836. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Guan, J.; Xu, Y.; Ren, H.; Jiang, J.; Wudu, M.; Wang, Q.; Su, H.; Zhang, Y.; Zhang, B.; et al. Silencing of CASC8 inhibits non-small cell lung cancer cells function and promotes sensitivity to osimertinib via FOXM1. J. Cancer 2021, 12, 387–396. [Google Scholar] [CrossRef]
- Yao, K.; Hua, L.; Wei, L.; Meng, J.; Hu, J. Correlation Between CASC8, SMAD7 Polymorphisms and the Susceptibility to Colorectal Cancer: An Updated Meta-Analysis Based on GWAS Results. Medicine 2015, 94, e1884. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Wang, Y.; Li, X.; Xiao, Y.; Wang, W. High Cancer Susceptibility Candidate 8 Expression Is Associated With Poor Prognosis of Pancreatic Adenocarcinoma: Validated Analysis Based on Four Cancer Databases. Front. Cell Dev. Biol. 2020, 8, 392. [Google Scholar] [CrossRef]
- Ji, J.; Xu, R.; Ding, K.; Bao, G.; Zhang, X.; Huang, B.; Wang, J. Long Noncoding RNA SChLAP1 Forms a Growth-Promoting Complex with HNRNPL in Human Glioblastoma through Stabilization of ACTN4 and Activation of NF-kappaB Signaling. Clin. Cancer Res. 2019, 25, 6868–6881. [Google Scholar] [CrossRef] [Green Version]
- Xiu, B.; Chi, Y.; Liu, L.; Chi, W.; Zhang, Q.; Chen, J.; Guo, R.; Si, J.; Li, L.; Xue, J.; et al. LINC02273 drives breast cancer metastasis by epigenetically increasing AGR2 transcription. Mol. Cancer 2019, 18, 187. [Google Scholar] [CrossRef] [Green Version]
- Fei, T.; Chen, Y.; Xiao, T.; Li, W.; Cato, L.; Zhang, P.; Cotter, M.B.; Bowden, M.; Lis, R.T.; Zhao, S.G.; et al. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc. Natl. Acad. Sci. USA 2017, 114, E5207–E5215. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; He, W.; Huang, J.; Wang, B.; Li, H.; Cai, Q.; Su, F.; Bi, J.; Liu, H.; Zhang, B.; et al. LNMAT1 promotes lymphatic metastasis of bladder cancer via CCL2 dependent macrophage recruitment. Nat. Commun. 2018, 9, 3826. [Google Scholar] [CrossRef] [PubMed]
- Listerman, I.; Sun, J.; Gazzaniga, F.S.; Lukas, J.L.; Blackburn, E.H. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res 2013, 73, 2817–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Z.; Gao, M.; Yin, Z.; Yan, L.; Cui, L. Association between lncRNA CASC8 polymorphisms and the risk of cancer: A meta-analysis. Cancer Manag. Res. 2018, 10, 3141–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Chen, S.H.; Lv, Q.L.; Sun, B.; Qu, Q.; Qin, C.Z.; Fan, L.; Guo, Y.; Cheng, L.; Zhou, H.H. Clinical Significance of Long Non-Coding RNA CASC8 rs10505477 Polymorphism in Lung Cancer Susceptibility, Platinum-Based Chemotherapy Response, and Toxicity. Int. J. Environ. Res. Public Health 2016, 13, 545. [Google Scholar] [CrossRef] [PubMed]
- Haerian, M.S.; Haerian, B.S.; Molanaei, S.; Kosari, F.; Sabeti, S.; Bidari-Zerehpoosh, F.; Abdolali, E. Lack of association of CASC8 rs1447295 with colorectal cancer in Iranian population: A multicenter case-control study. Gene 2017, 634, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Kastler, S.; Honold, L.; Luedeke, M.; Kuefer, R.; Moller, P.; Hoegel, J.; Vogel, W.; Maier, C.; Assum, G. POU5F1P1, a putative cancer susceptibility gene, is overexpressed in prostatic carcinoma. Prostate 2010, 70, 666–674. [Google Scholar] [CrossRef]
- Ma, G.; Gu, D.; Lv, C.; Chu, H.; Xu, Z.; Tong, N.; Wang, M.; Tang, C.; Xu, Y.; Zhang, Z.; et al. Genetic variant in 8q24 is associated with prognosis for gastric cancer in a Chinese population. J. Gastroenterol. Hepatol. 2015, 30, 689–695. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Uleri, E.; Caglioti, C.; Dolei, A. Type I IFN family members: Similarity, differences and interaction. Cytokine Growth Factor Rev. 2015, 26, 103–111. [Google Scholar] [CrossRef]
- Broggi, A.; Granucci, F.; Zanoni, I. Type III interferons: Balancing tissue tolerance and resistance to pathogen invasion. J. Exp. Med. 2020, 217, e20190295. [Google Scholar] [CrossRef]
- Burke, J.D.; Young, H.A. IFN-γ: A cytokine at the right time, is in the right place. Semin. Immunol. 2019, 43, 101280. [Google Scholar] [CrossRef]
- Hu, X.; Ivashkiv, L.B. Cross-regulation of signaling pathways by interferon-γ: Implications for immune responses and autoimmune diseases. Immunity 2009, 31, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Christopoulos, P.F.; Halder, S.; Lunde, A.; Beraki, K.; Speth, M.; Corthay, A. Toll-Like Receptor Ligands and Interferon-γ Synergize for Induction of Antitumor M1 Macrophages. Front. Immunol. 2017, 8, 1383. [Google Scholar] [CrossRef] [PubMed]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-γ at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Chhatar, S.; Mishra, A.; Lal, G. Natural killer T cell activation increases iNOS(+)CD206(-) M1 macrophage and controls the growth of solid tumor. J. Immunother. Cancer 2019, 7, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, M.G.; Jacobs, T.; Lamarca, A.; Hubner, R.A.; Valle, J.W.; Amir, E. Impact of high tumor mutational burden in solid tumors and challenges for biomarker application. Cancer Treat. Rev. 2020, 89, 102084. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dreicer, R. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (N. Y., NY) 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Chi, H.; Gou, S.; Guo, X.; Li, L.; Peng, G.; Zhang, J.; Xu, J.; Nian, S.; Yuan, Q. An Aggrephagy-Related LncRNA Signature for the Prognosis of Pancreatic Adenocarcinoma. Genes 2023, 14, 124. https://doi.org/10.3390/genes14010124
Huang X, Chi H, Gou S, Guo X, Li L, Peng G, Zhang J, Xu J, Nian S, Yuan Q. An Aggrephagy-Related LncRNA Signature for the Prognosis of Pancreatic Adenocarcinoma. Genes. 2023; 14(1):124. https://doi.org/10.3390/genes14010124
Chicago/Turabian StyleHuang, Xueyuan, Hao Chi, Siqi Gou, Xiyuan Guo, Lin Li, Gaoge Peng, Jinhao Zhang, Jiayu Xu, Siji Nian, and Qing Yuan. 2023. "An Aggrephagy-Related LncRNA Signature for the Prognosis of Pancreatic Adenocarcinoma" Genes 14, no. 1: 124. https://doi.org/10.3390/genes14010124
APA StyleHuang, X., Chi, H., Gou, S., Guo, X., Li, L., Peng, G., Zhang, J., Xu, J., Nian, S., & Yuan, Q. (2023). An Aggrephagy-Related LncRNA Signature for the Prognosis of Pancreatic Adenocarcinoma. Genes, 14(1), 124. https://doi.org/10.3390/genes14010124