
The Two-Faced Role of crAssphage Subfamilies in Obesity and Metabolic Syndrome: Between Good and Evil
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Acquisition of Viral Data and Mapping to the crAssphage Genome Database
2.2. crAssphages α- and β-Diversities
2.3. Association of crAssphage Abundance with Metadata Variables and Microbiota
2.4. Correlation of crAssphage Abundance with Their Bacterial Host
3. Results
3.1. Decreased Abundance and Prevalence of crAssphages Were Detected in the O and OMS Groups
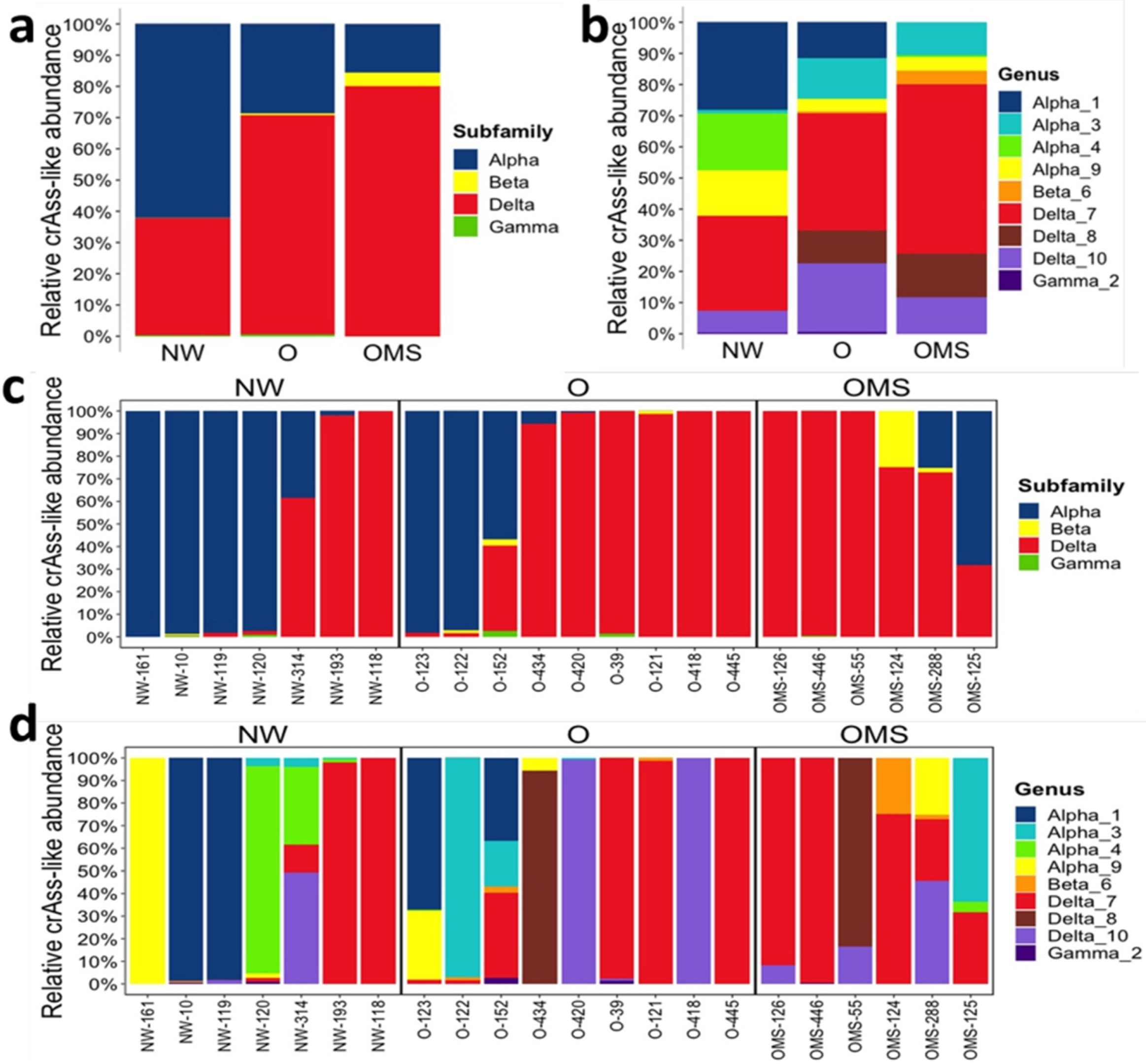
3.2. Changes in crAssphage Taxonomy Characterize the O and OMS Groups
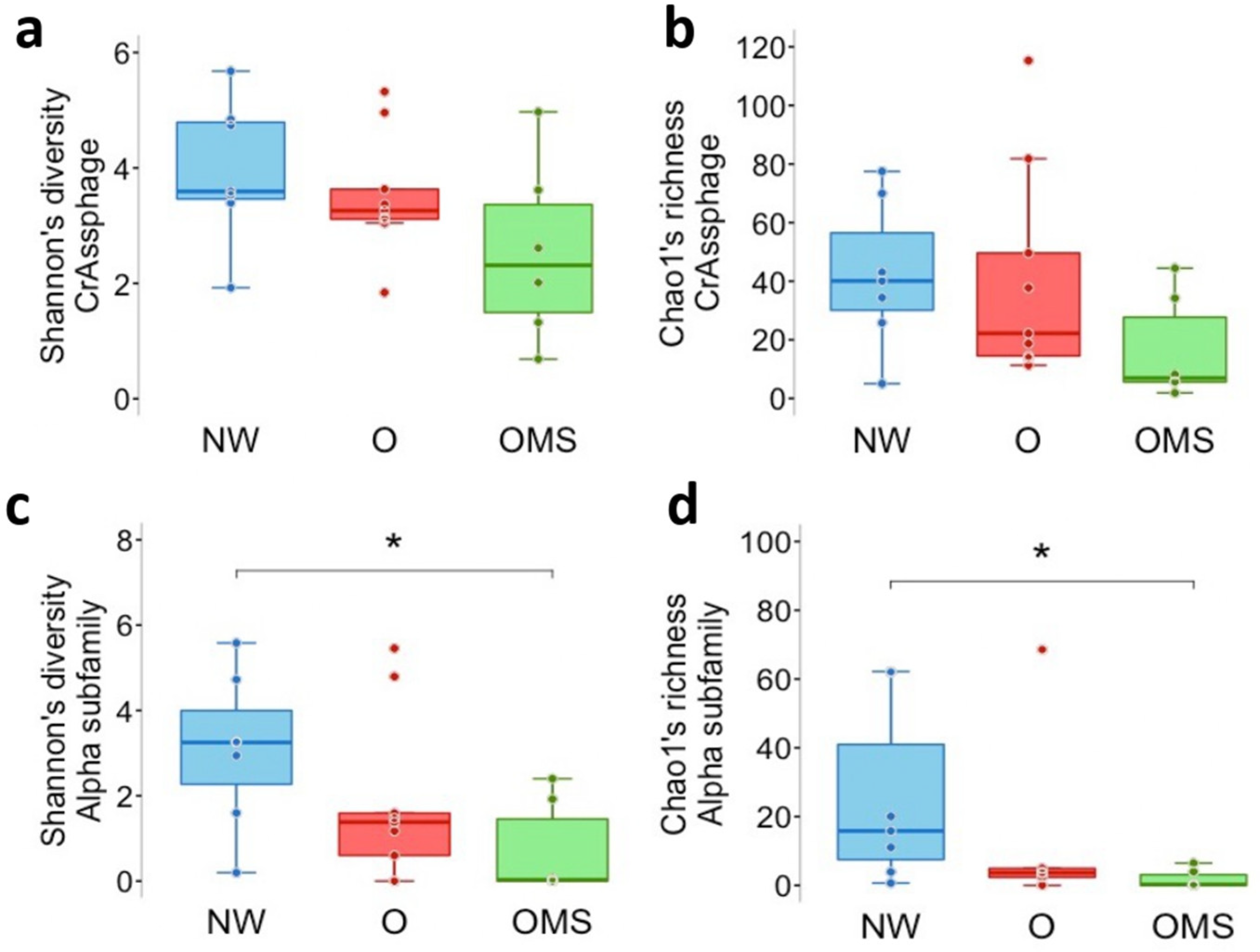
3.3. Decreased Diversity and Richness of α crAssphages Are Associated with the O and OMS Groups
3.4. Host Bacteria of crAssphages Were Decreased in the O and OMS Groups
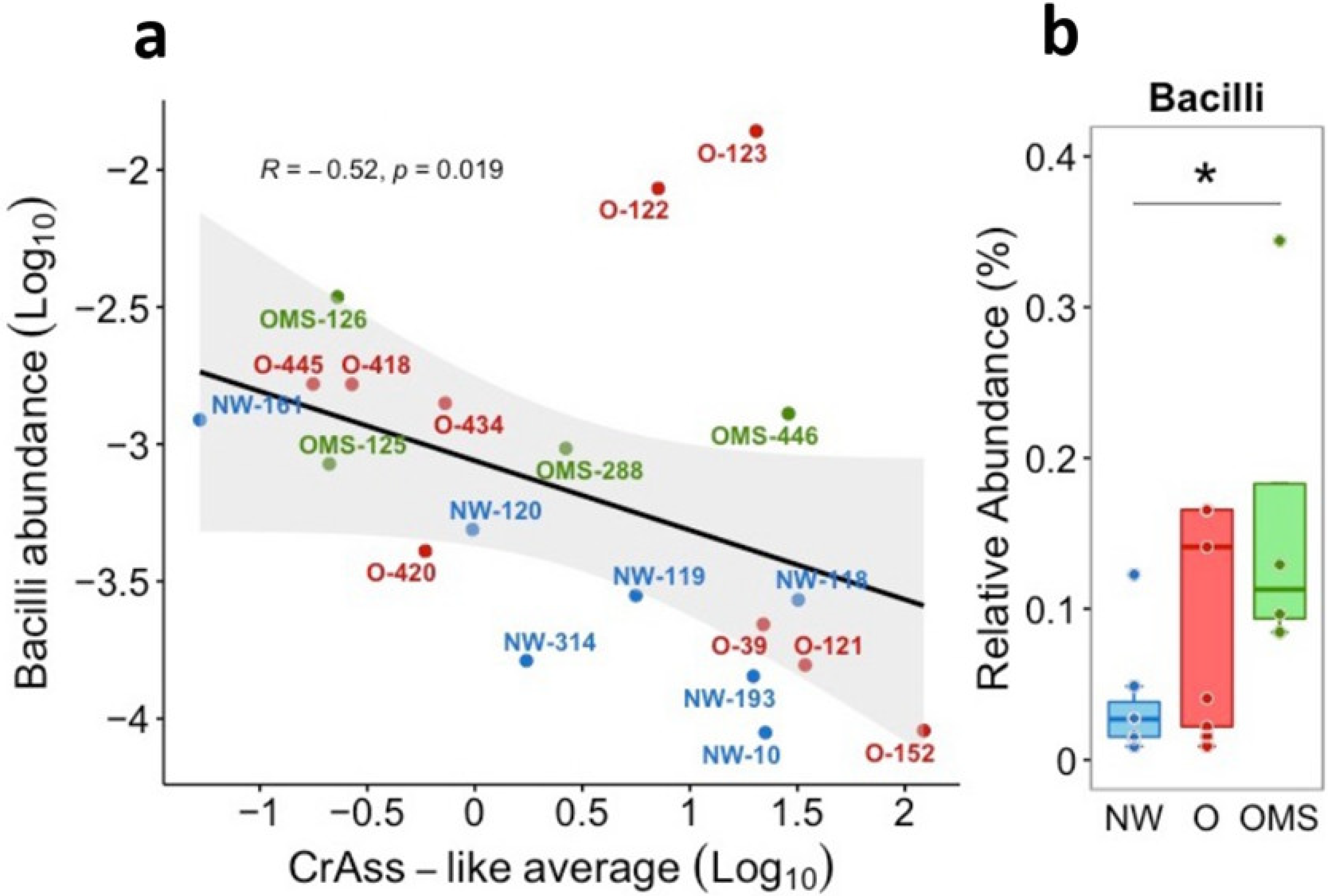
3.5. Increased Bacilli Were Associated with Decreased crAssphage Abundance in OMS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bikel, S.; Gallardo-Becerra, L.; Cornejo-Granados, F.; Ochoa-Leyva, A. Protocol for the isolation, sequencing, and analysis of the gut phageome from human fecal samples. STAR Protoc. 2022, 3, 101–170. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe 2019, 26, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.; Zhao, R.; Chan, P.K.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.; et al. Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe 2016, 19, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; You, X.; Mai, G.; Tokuyasu, T.; Liu, C. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome 2018, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, A.; Blanton, L.V.; Cao, S.; Zhao, G.; Manary, M.; Trehan, I.; Smith, M.I.; Wang, D.; Herbert, W.V.; Rohwer, F.; et al. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA 2015, 112, 11941–11946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikel, S.; López-Leal, G.; Cornejo-Granados, F.; Gallardo-Becerra, L.; García-López, R.; Sánchez, F.; Equihua-Medina, E.; Ochoa-Romo, J.P.; López-Contreras, B.E.; Canizales-Quintero, S.; et al. Gut dsDNA virome shows diversity and richness alterations associated with childhood obesity and metabolic syndrome. iScience 2021, 24, 102900. [Google Scholar] [CrossRef]
- Rossi, A.; Treu, L.; Toppo, S.; Zschach, H.; Campanaro, S.; Dutilh, B.E. Evolutionary study of the crassphage virus at gene level. Viruses 2020, 12, 1035. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Konnin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Echeverría, M.; Equihua-Medina, E.; Cornejo-Granados, F.; Hernández-Reyna, A.; Sánchez, F.; López-Contreras, B.E.; Canizales-Quintero, S.; Ochoa-Leyva, A. Whole-genome of Mexican-crAssphage isolated from the human gut microbiome. BMC Res. Notes 2018, 11, 902. [Google Scholar] [CrossRef] [Green Version]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and taxonomy of crAss-like bacteriophages, the most abundant virus in the human gut. Cell Host Microbe 2018, 24, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.Y.; Zhang, W.; Tong, Y.G.; Chen, S.P. crAssphage is not associated with diarrhoea and has high genetic diversity. Epidemiol. Infect. 2016, 144, 3549–3553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockdale, S.R.; Hill, C. Progress and prospects of the healthy human gut virome. Curr. Opin. Virol. 2021, 51, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host Microbe 2020, 28, 724–740. [Google Scholar] [CrossRef] [PubMed]
- Siranosian, B.A.; Tamburini, F.B.; Sherlock, G.; Bhatt, A.S. Acquisition, transmission and strain diversity of human gut-colonizing crAss-like phages. Nat. Commun. 2020, 11, 280. [Google Scholar] [CrossRef] [Green Version]
- Cinek, O.; Mazankova, K.; Kramna, L.; Odeh, R.; Alassaf, A.; Ibekwe, M.U.; Ahmadov, G.; Mekki, H.; Abdullah, M.A.; Elmahi, B.M.E.; et al. Quantitative CrAssphage real-time PCR assay derived from data of multiple geographically distant populations. J. Med. Virol. 2018, 90, 767–771. [Google Scholar] [CrossRef]
- Stachler, E.; Kelty, C.; Sivaganesan, M.; Li, X.; Bibby, K.; Shanks, O.C. Quantitative crAssphage PCR assays for human fecal pollution measurement. Environ. Sci. Technol. 2017, 51, 9146–9154. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honap, T.P.; Sankaranarayanan, K.; Schnorr, S.L.; Ozga, A.T.; Warinner, C.; Lewis, C.M. Biogeographic study of human gut-associated crAssphage suggests impacts from industrialization and recent expansion. PLoS ONE 2020, 15, 0226930. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Zhu, Y.; Kong, C.; Xia, K.; Li, H.; Zhu, Y.; Zhang, X.; Liu, Y.; Zhong, H.; Yang, R.; et al. Alterations, interactions, and diagnostic potential of gut bacteria and viruses in colorectal cancer. Front. Cell Infect. Microbiol. 2021, 11, 657867. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gan, D.; Li, Y.; Wang, X.; Jin, L.; Qin, K.; Cui, C.; Wu, J.; Wang, Z. Quyushengxin formula causes differences in bacterial and phage composition in ulcerative colitis patients. Evid. Based Complement. Alternat. Med. 2020, 2020, 5859023. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Becerra, L.; Cornejo-Granados, F.; García-López, R.; Valdez-Lara, A.; Bikel, S.; Canizales-Quinteros, S.; López-Contreras, B.E.; Mendoza-Vargas, A.; Nielsen, H.; Ochoa-Leyva, A. Metatranscriptomic analysis to define the Secrebiome, and 16S rRNA profiling of the gut microbiome in obesity and metabolic syndrome of Mexican children. Microb. Cell Fact. 2020, 19, 61. [Google Scholar] [CrossRef] [Green Version]
- Guerin, E.; Shkoporov, A.N.; Stockdale, S.R.; Comas, J.C.; Khoklova, E.V.; Clooney, A.G.; Daly, K.M.; Draper, L.A.; Niamh, S.; Scholz, D.; et al. Isolation and characterisation of ΦcrAss002, a crAss-like phage from the human gut that infects Bacteroides xylanisolvens. Microbiome 2021, 9, 89. [Google Scholar] [CrossRef]
- Hossein, B.B.; Behrouz, N.; Dasiush, S.; Hamed, E.L. Evaluation of a human gut-associated phage and gut dominant microbial phyla in the metabolic syndrome. Clin. Nutr. 2022, 50, 133–137. [Google Scholar] [CrossRef]
- De Jonge, P.A.; Wortelboer, K.; Scheithauer, T.P.M.; van den Born, B.-J.H.; Zwinderman, A.H.; Nobrega, F.L.; Dutilh, B.E.; Niewwdrop, M.; Herrema, H. Gut virome profiling identifies a widespread bacteriophage family associated with metabolic syndrome. Nat. Commun. 2022, 13, 3594. [Google Scholar] [CrossRef]
- Draper, L.A.; Ryan, F.J.; Smith, M.K.; Jalanka, J.; Mattila, E.; Arkkila, P.A.; Ross, R.P.; Satokari, R.; Hill, C. Long-term colonisation with donor bacteriophages following successful faecal microbial transplantation. Microbiome 2018, 6, 220. [Google Scholar] [CrossRef]
- Febvre, H.P.; Rao, S.; Gindin, M.; Goodwin, N.D.M.; Finer, E.; Vivanco, J.S.; Lu, S.; Manter, D.K.; Wallace, T.C.; Weir, T.L. PHAGE study: Effects of supplemental bacteriophage intake on inflammation and gut microbiota in healthy adults. Nutrients 2019, 11, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gindin, M.; Febvre, H.P.; Rao, S.; Wallace, T.C.; Weir, T.L. Bacteriophage for Gastrointestinal Health (PHAGE) Study: Evaluating the Safety and Tolerability of Supplemental Bacteriophage Consumption. J. Am. Coll. Nutr. 2019, 38, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.A.; Shapiro, J.A.; Church, T.R.; Miller, G.; Trinh-Shevrin, C.; Yuen, E.; Friedlander, C.; Hayes, R.B.; Ahn, J. A taxonomic signature of obesity in a large study of American adults. Sci. Rep. 2018, 8, 9749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinek, O.; Kramna, L.; Lin, J.; Oikarinen, S.; Kolarova, K.; Ilonen, J.; Simell, O.; Veijola, R.; Autio, R.; Hyöty, H. Imbalance of bacteriome profiles within the Finnish Diabetes Prediction and Prevention study: Parallel use of 16S profiling and virome sequencing in stool samples from children with islet autoimmunity and matched controls. Pediatr. Diabetes 2017, 18, 588–598. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cervantes-Echeverría, M.; Gallardo-Becerra, L.; Cornejo-Granados, F.; Ochoa-Leyva, A. The Two-Faced Role of crAssphage Subfamilies in Obesity and Metabolic Syndrome: Between Good and Evil. Genes 2023, 14, 139. https://doi.org/10.3390/genes14010139
Cervantes-Echeverría M, Gallardo-Becerra L, Cornejo-Granados F, Ochoa-Leyva A. The Two-Faced Role of crAssphage Subfamilies in Obesity and Metabolic Syndrome: Between Good and Evil. Genes. 2023; 14(1):139. https://doi.org/10.3390/genes14010139
Chicago/Turabian StyleCervantes-Echeverría, Melany, Luigui Gallardo-Becerra, Fernanda Cornejo-Granados, and Adrian Ochoa-Leyva. 2023. "The Two-Faced Role of crAssphage Subfamilies in Obesity and Metabolic Syndrome: Between Good and Evil" Genes 14, no. 1: 139. https://doi.org/10.3390/genes14010139