
Prenatal Screening and Diagnostic Considerations for 22q11.2 Microdeletions

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Methods
2.1. Literature Search
2.2. Inclusion and Exclusion Criteria
2.3. Data Extraction and Synthesis
3. Results
Review and Recommendations
3.1.1. Diagnostic Testing for 22q11.2 Microdeletion in Pregnancy
3.1.2. Noninvasive Prenatal Screening (NIPS) for 22q11.2 Microdeletions
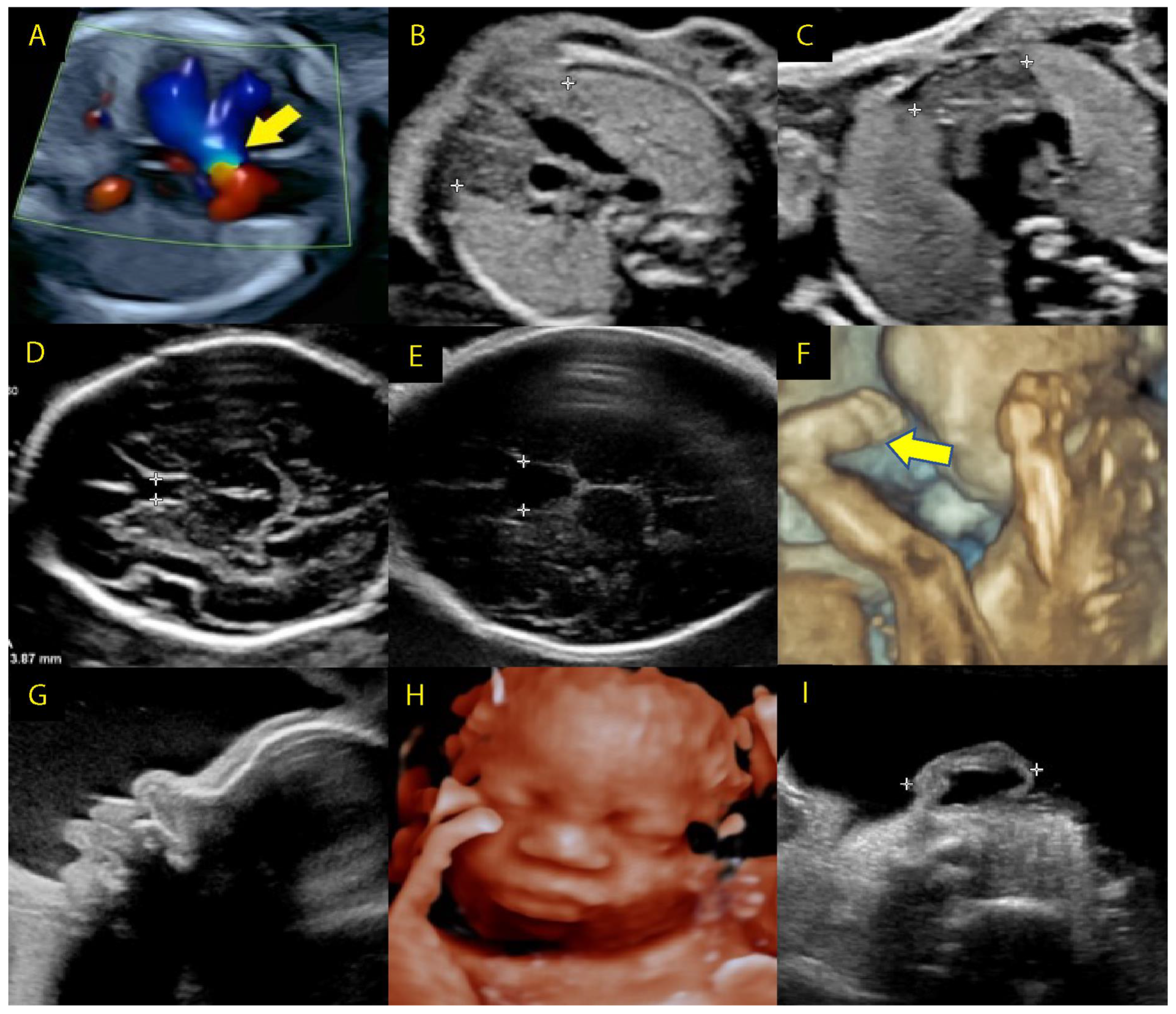
3.1.3. Prenatal Screening—Ultrasound Imaging in 22q11.2 Microdeletions
3.1.4. First-Trimester Ultrasound
3.1.5. Second-Trimester Ultrasound
3.1.6. Third-Trimester Ultrasound
3.1.7. Prenatal Screening—MRI Imaging in 22q11.2 Microdeletion
3.1.8. Reproductive Options for a Parent with 22q11.2 Microdeletion
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagojevic, C.; Heung, T.; Theriault, M.; Tomita-Mitchell, A.; Chakraborty, P.; Kernohan, K.; Bulman, D.E.; Bassett, A.S. Estimate of the Contemporary Live-Birth Prevalence of Recurrent 22q11.2 Deletions: A Cross-Sectional Analysis from Population-Based Newborn Screening. CMAJ Open 2021, 9, E802–E809. [Google Scholar] [CrossRef] [PubMed]
- Grati, F.R.; Molina Gomes, D.; Ferreira, J.C.P.B.; Dupont, C.; Alesi, V.; Gouas, L.; Horelli-Kuitunen, N.; Choy, K.W.; García-Herrero, S.; de la Vega, A.G.; et al. Prevalence of Recurrent Pathogenic Microdeletions and Microduplications in over 9500 Pregnancies. Prenat. Diagn. 2015, 35, 801–809. [Google Scholar] [CrossRef]
- Maisenbacher, M.K.; Merrion, K.; Pettersen, B.; Young, M.; Paik, K.; Iyengar, S.; Kareht, S.; Sigurjonsson, S.; Demko, Z.P.; Martin, K.A. Incidence of the 22q11.2 Deletion in a Large Cohort of Miscarriage Samples. Mol. Cytogenet. 2017, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalczyk, K.; Bartnik-Głaska, M.; Smyk, M.; Plaskota, I.; Bernaciak, J.; Kędzior, M.; Wiśniowiecka-Kowalnik, B.; Jakubów-Durska, K.; Braun-Walicka, N.; Barczyk, A.; et al. Prenatal Diagnosis by Array Comparative Genomic Hybridization in Fetuses with Cardiac Abnormalities. Genes 2021, 12, 2021. [Google Scholar] [CrossRef] [PubMed]
- Noël, A.-C.; Pelluard, F.; Delezoide, A.-L.; Devisme, L.; Loeuillet, L.; Leroy, B.; Martin, A.; Bouvier, R.; Laquerriere, A.; Jeanne-Pasquier, C.; et al. Fetal Phenotype Associated with the 22q11 Deletion. Am. J. Med. Genet. Part A 2014, 164, 2724–2731. [Google Scholar] [CrossRef]
- Bretelle, F.; Beyer, L.; Pellissier, M.C.; Missirian, C.; Sigaudy, S.; Gamerre, M.; D’Ercole, C.; Philip, N. Prenatal and Postnatal Diagnosis of 22q11.2 Deletion Syndrome. Eur. J. Med. Genet. 2010, 53, 367–370. [Google Scholar] [CrossRef]
- Lamouroux, A.; Mousty, E.; Prodhomme, O.; Bigi, N.; Le Gac, M.-P.; Letouzey, V.; De Tayrac, R.; Mares, P. Absent or hypoplastic thymus: A marker for 22q11.2 microdeletion syndrome in case of polyhydramnios. J. Gynecol. Obs. Biol. Reprod. 2016, 45, 388–396. [Google Scholar] [CrossRef]
- Palmer, L.D.; Butcher, N.J.; Boot, E.; Hodgkinson, K.A.; Heung, T.; Chow, E.W.C.; Guna, A.; Crowley, T.B.; Zackai, E.; McDonald-McGinn, D.M.; et al. Elucidating the Diagnostic Odyssey of 22q11.2 Deletion Syndrome. Am. J. Med. Genet. A 2018, 176, 936–944. [Google Scholar] [CrossRef]
- Campbell, I.M.; Sheppard, S.E.; Crowley, T.B.; McGinn, D.E.; Bailey, A.; McGinn, M.J.; Unolt, M.; Homans, J.F.; Chen, E.Y.; Salmons, H.I.; et al. What Is New with 22q? An Update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am. J. Med. Genet. A 2018, 176, 2058–2069. [Google Scholar] [CrossRef]
- Sarac Sivrikoz, T.; Basaran, S.; Has, R.; Karaman, B.; Kalelioglu, I.H.; Kirgiz, M.; Altunoglu, U.; Yuksel, A. Prenatal Sonographic and Cytogenetic/Molecular Findings of 22q11.2 Microdeletion Syndrome in 48 Confirmed Cases in a Single Tertiary Center. Arch. Gynecol. Obs. 2022, 305, 323–342. [Google Scholar] [CrossRef]
- Swillen, A.; Moss, E.; Duijff, S. Neurodevelopmental Outcome in 22q11.2 Deletion Syndrome and Management. Am. J. Med. Genet. A 2018, 176, 2160–2166. [Google Scholar] [CrossRef] [PubMed]
- Solot, C.B.; Sell, D.; Mayne, A.; Baylis, A.L.; Persson, C.; Jackson, O.; McDonald-McGinn, D.M. Speech-Language Disorders in 22q11.2 Deletion Syndrome: Best Practices for Diagnosis and Management. Am. J. Speech-Lang. Pathol. 2019, 28, 984–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskarsdottir, S.; Boot, E.; Crowley, T.B.; Loo, J.C.Y.; Arganbright, J.; Armando, M.; Baylis, A.L.; Breetvelt, E.J.; Castelein, R.M.; Chadehumbe, M.A.; et al. Updated Clinical Practice Recommendations for Managing Children with 22q11.2 Deletion Syndrome. Genet. Med. 2022, in press. [Google Scholar]
- Swillen, A. The Importance of Understanding Cognitive Trajectories: The Case of 22q11.2 Deletion Syndrome. Curr. Opin. Psychiatry 2016, 29, 133–137. [Google Scholar] [CrossRef]
- Fung, W.L.A.; Butcher, N.J.; Costain, G.; Andrade, D.M.; Boot, E.; Chow, E.W.C.; Chung, B.; Cytrynbaum, C.; Faghfoury, H.; Fishman, L.; et al. Practical Guidelines for Managing Adults with 22q11.2 Deletion Syndrome. Genet. Med. 2015, 17, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Boot, E.; Oskarsdottir, S.; Loo, J.C.Y.; Crowley, T.B.; Andrade, D.M.; Arganbright, J.; Castelein, R.M.; Cserti-Gazdewich, C.; de Reuver, S.; Fiksinski, A.M.; et al. Updated Clinical Practice Recommendations for Managing Adults with 22q11.2 Deletion Syndrome. Genet. Med. 2022, in press. [Google Scholar]
- Shaikh, T.H.; Kurahashi, H.; Saitta, S.C.; O’Hare, A.M.; Hu, P.; Roe, B.A.; Driscoll, D.A.; McDonald-McGinn, D.M.; Zackai, E.H.; Budarf, M.L.; et al. Chromosome 22-Specific Low Copy Repeats and the 22q11.2 Deletion Syndrome: Genomic Organization and Deletion Endpoint Analysis. Hum. Mol. Genet. 2000, 9, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Mikhail, F.M.; Burnside, R.D.; Rush, B.; Ibrahim, J.; Godshalk, R.; Rutledge, S.L.; Robin, N.H.; Descartes, M.D.; Carroll, A.J. The Recurrent Distal 22q11.2 Microdeletions Are Often de Novo and Do Not Represent a Single Clinical Entity: A Proposed Categorization System. Genet. Med. 2014, 16, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Racedo, S.E.; McDonald-McGinn, D.M.; Chung, J.H.; Goldmuntz, E.; Zackai, E.; Emanuel, B.S.; Zhou, B.; Funke, B.; Morrow, B.E. Mouse and Human CRKL Is Dosage Sensitive for Cardiac Outflow Tract Formation. Am. J. Hum. Genet. 2015, 96, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Rivera, E.; Liu, Y.P.; Verbitsky, M.; Anderson, B.R.; Capone, V.P.; Otto, E.A.; Yan, Z.; Mitrotti, A.; Martino, J.; Steers, N.J.; et al. Genetic Drivers of Kidney Defects in the DiGeorge Syndrome. N. Engl. J. Med. 2017, 376, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, T.; Niihori, T.; Tanaka, N.; Kawai, M.; Nagashima, T.; Funayama, R.; Nakayama, K.; Nakashima, S.; Kato, F.; Fukami, M.; et al. TBX1 Mutation Identified by Exome Sequencing in a Japanese Family with 22q11.2 Deletion Syndrome-like Craniofacial Features and Hypocalcemia. PLoS ONE 2014, 9, e91598. [Google Scholar] [CrossRef] [PubMed]
- Akolekar, R.; Beta, J.; Picciarelli, G.; Ogilvie, C.; D’Antonio, F. Procedure-Related Risk of Miscarriage Following Amniocentesis and Chorionic Villus Sampling: A Systematic Review and Meta-Analysis. Ultrasound Obstet. Gynecol. 2015, 45, 16–26. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 200: Early Pregnancy Loss. Obstet. Gynecol. 2018, 132, e197. [Google Scholar] [CrossRef]
- Dungan, J.S.; Klugman, S.; Darilek, S.; Malinowski, J.; Akkari, Y.M.N.; Monaghan, K.G.; Erwin, A.; Best, R.G. Noninvasive Prenatal Screening (NIPS) for Fetal Chromosome Abnormalities in a General-Risk Population: An Evidence-Based Clinical Guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2022. [Google Scholar] [CrossRef]
- Dar, P.; Jacobsson, B.; Clifton, R.; Egbert, M.; Malone, F.; Wapner, R.J.; Roman, A.S.; Khalil, A.; Faro, R.; Madankumar, R.; et al. Cell-Free DNA Screening for Prenatal Detection of 22q11.2 Deletion Syndrome. Am. J. Obstet. Gynecol. 2022, 227, 79.e1–79.e11. [Google Scholar] [CrossRef]
- Rose, N.C.; Barrie, E.S.; Malinowski, J.; Jenkins, G.P.; McClain, M.R.; LaGrave, D.; Leung, M.L.; ACMG Professional Practice and Guidelines Committee. Electronic address: Documents@acmg.net Systematic Evidence-Based Review: The Application of Noninvasive Prenatal Screening Using Cell-Free DNA in General-Risk Pregnancies. Genet. Med. 2022, 24, 1379–1391. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Hsieh, T.-T.; Cheng, P.-J.; Hung, T.-H.; Chan, K.-S.; Tsai, C.; Shaw, S.W. Taiwanese Clinical Experience with Noninvasive Prenatal Testing for DiGeorge Syndrome. FDT 2021, 48, 672–677. [Google Scholar] [CrossRef]
- Bevilacqua, E.; Jani, J.C.; Chaoui, R.; Suk, E.-K.A.; Palma-Dias, R.; Ko, T.-M.; Warsof, S.; Stokowski, R.; Jones, K.J.; Grati, F.R.; et al. Performance of a Targeted Cell-Free DNA Prenatal Test for 22q11.2 Deletion in a Large Clinical Cohort. Ultrasound Obstet. Gynecol. 2021, 58, 597–602. [Google Scholar] [CrossRef]
- Kagan, K.O.; Hoopmann, M.; Pfaff, T.; Prodan, N.; Wagner, P.; Schmid, M.; Dufke, A.; Mau-Holzmann, U.; Brucker, S.; Marcato, L.; et al. First Trimester Screening for Common Trisomies and Microdeletion 22q11.2 Syndrome Using Cell-Free DNA: A Prospective Clinical Study. FDT 2020, 47, 841–852. [Google Scholar] [CrossRef]
- Martin, K.; Iyengar, S.; Kalyan, A.; Lan, C.; Simon, A.L.; Stosic, M.; Kobara, K.; Ravi, H.; Truong, T.; Ryan, A.; et al. Clinical Experience with a Single-Nucleotide Polymorphism-Based Non-Invasive Prenatal Test for Five Clinically Significant Microdeletions. Clin. Genet. 2018, 93, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, L.D.; McManus, Z.; Heung, T.; McAlpine, G.; Blagojevic, C.; Corral, M.; Bassett, A.S. Reproductive Outcomes in Adults with 22q11.2 Deletion Syndrome. Genes 2022, 13, 2126. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.D.; Heung, T.; Corral, M.; Boot, E.; Brooks, S.G.; Bassett, A.S. Sexual Knowledge and Behaviour in 22q11.2 Deletion Syndrome, a Complex Care Condition. J. Appl. Res. Intellect. Disabil. 2022, 35, 966–975. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Simpson, J.L.; Jackson, L.G.; Elias, S.; Holzgreve, W.; Evans, M.I.; Dukes, K.A.; Sullivan, L.M.; Klinger, K.W.; Bischoff, F.Z.; et al. Fetal Gender and Aneuploidy Detection Using Fetal Cells in Maternal Blood: Analysis of NIFTY I Data. Prenat. Diagn. 2002, 22, 609–615. [Google Scholar] [CrossRef]
- Vossaert, L.; Chakchouk, I.; Zemet, R.; Van den Veyver, I.B. Overview and Recent Developments in Cell-Based Noninvasive Prenatal Testing. Prenat. Diagn. 2021, 41, 1202–1214. [Google Scholar] [CrossRef]
- Choolani, M.; Mahyuddin, A.P.; Hahn, S. The Promise of Fetal Cells in Maternal Blood. Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 655–667. [Google Scholar] [CrossRef]
- Schmorl, G. Pathologisch-Anatomische Untersuchungen Über Puerperal-Eklampsie; F.C.W. Vogel: Leipzig, Germany, 1893. [Google Scholar]
- Jing, X.-Y.; Zhang, Y.-L.; Zhen, L.; Li, Y.-L.; Li, D.-Z. Prenatal Sonographic Findings in a Cohort of Foetuses with a Confirmed 22q11.2 Microdeletion at a Single Chinese Tertiary Centre. J. Obstet. Gynaecol. 2022, 42, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Besseau-Ayasse, J.; Violle-Poirsier, C.; Bazin, A.; Gruchy, N.; Moncla, A.; Girard, F.; Till, M.; Mugneret, F.; Coussement, A.; Pelluard, F.; et al. A French Collaborative Survey of 272 Fetuses with 22q11.2 Deletion: Ultrasound Findings, Fetal Autopsies and Pregnancy Outcomes. Prenat. Diagn. 2014, 34, 424–430. [Google Scholar] [CrossRef]
- Li, S.; Han, X.; Ye, M.; Chen, S.; Shen, Y.; Niu, J.; Wang, Y.; Xu, C. Prenatal Diagnosis of Microdeletions or Microduplications in the Proximal, Central, and Distal Regions of Chromosome 22q11.2: Ultrasound Findings and Pregnancy Outcome. Front. Genet. 2019, 10, 813. [Google Scholar] [CrossRef] [Green Version]
- Schindewolf, E.; Khalek, N.; Johnson, M.P.; Gebb, J.; Coleman, B.; Crowley, T.B.; Zackai, E.H.; McDonald-McGinn, D.M.; Moldenhauer, J.S. Expanding the Fetal Phenotype: Prenatal Sonographic Findings and Perinatal Outcomes in a Cohort of Patients with a Confirmed 22q11.2 Deletion Syndrome. Am. J. Med. Genet. Part A 2018, 176, 1735–1741. [Google Scholar] [CrossRef]
- Fagerberg, C.R.; Graakjaer, J.; Heinl, U.D.; Ousager, L.B.; Dreyer, I.; Kirchhoff, M.; Rasmussen, A.A.; Lautrup, C.K.; Birkebaek, N.; Sorensen, K. Heart Defects and Other Features of the 22q11 Distal Deletion Syndrome. Eur. J. Med. Genet. 2013, 56, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.; Jansen, F.A.R.; Blumenfeld, Y.J.; Fisher, A.; Odibo, A.O.; Haak, M.C.; Borrell, A. Genomic Microarray in Fetuses with Increased Nuchal Translucency and Normal Karyotype: A Systematic Review and Meta-Analysis. Ultrasound Obs. Gynecol. 2015, 46, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Karim, J.N.; Bradburn, E.; Roberts, N.; Papageorghiou, A.T.; ACCEPTS Study. First-Trimester Ultrasound Detection of Fetal Heart Anomalies: Systematic Review and Meta-Analysis. Ultrasound Obstet. Gynecol. 2022, 59, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Karim, J.N.; Roberts, N.W.; Salomon, L.J.; Papageorghiou, A.T. Systematic Review of First-Trimester Ultrasound Screening for Detection of Fetal Structural Anomalies and Factors That Affect Screening Performance. Ultrasound Obstet. Gynecol. 2017, 50, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Pauta, M.; Grande, M.; Rodriguez-Revenga, L.; Kolomietz, E.; Borrell, A. Added Value of Chromosomal Microarray Analysis over Karyotyping in Early Pregnancy Loss: Systematic Review and Meta-Analysis. Ultrasound Obstet. Gynecol. 2018, 51, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Y.; Chen, Y.; Zhou, R.; Sang, Z.; Meng, L.; Tan, J.; Qiao, F.; Bao, Q.; Luo, D.; et al. Systematic Analysis of Copy-Number Variations Associated with Early Pregnancy Loss. Ultrasound Obstet. Gynecol. 2020, 55, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, K.; Bartnik-Głaska, M.; Smyk, M.; Plaskota, I.; Bernaciak, J.; Kędzior, M.; Wiśniowiecka-Kowalnik, B.; Deperas, M.; Domaradzka, J.; Łuszczek, A.; et al. Comparative Genomic Hybridization to Microarrays in Fetuses with High-Risk Prenatal Indications: Polish Experience with 7400 Pregnancies. Genes 2022, 13, 690. [Google Scholar] [CrossRef]
- Li, L.; Bahtiyar, M.O.; Buhimschi, C.S.; Zou, L.; Zhou, Q.-C.; Copel, J.A. Assessment of the Fetal Thymus by Two- and Three-Dimensional Ultrasound during Normal Human Gestation and in Fetuses with Congenital Heart Defects. Ultrasound Obstet. Gynecol. 2011, 37, 404–409. [Google Scholar] [CrossRef]
- Chaoui, R.; Kalache, K.D.; Heling, K.S.; Tennstedt, C.; Bommer, C.; Körner, H. Absent or Hypoplastic Thymus on Ultrasound: A Marker for Deletion 22q11.2 in Fetal Cardiac Defects. Ultrasound Obstet. Gynecol. 2002, 20, 546–552. [Google Scholar] [CrossRef]
- Chaoui, R.; Heling, K.-S.; Sarut Lopez, A.; Thiel, G.; Karl, K. The Thymic–Thoracic Ratio in Fetal Heart Defects: A Simple Way to Identify Fetuses at High Risk for Microdeletion 22q11. Ultrasound Obstet. Gynecol. 2011, 37, 397–403. [Google Scholar] [CrossRef]
- Dou, Y.; Schindewolf, E.; Crowley, T.B.; McDonald-McGinn, D.; Moldenhauer, J.S.; Coleman, B.; Oliver, E.R.; Sullivan, K.E. The Association of Fetal Thymus Size with Subsequent T Cell Counts in 22q11.2 Deletion Syndrome. J. Clin. Immunol. 2020, 40, 783–785. [Google Scholar] [CrossRef]
- Moore, J.W.; Binder, G.A.; Berry, R. Prenatal Diagnosis of Aneuploidy and Deletion 22q11.2 in Fetuses with Ultrasound Detection of Cardiac Defects. Am. J. Obstet. Gynecol. 2004, 191, 2068–2073. [Google Scholar] [CrossRef] [PubMed]
- Volpe, P.; Marasini, M.; Caruso, G.; Marzullo, A.; Buonadonna, A.L.; Arciprete, P.; Paolo, S.D.; Volpe, G.; Gentile, M. 22q11 Deletions in Fetuses with Malformations of the Outflow Tracts or Interruption of the Aortic Arch: Impact of Additional Ultrasound Signs. Prenat. Diagn. 2003, 23, 752–757. [Google Scholar] [CrossRef]
- Devriendt, K.; Moerman, P.; Schoubroeck, D.V.; Vandenberghe, K.; Fryns, J.P. Chromosome 22q11 Deletion Presenting as the Potter Sequence. J. Med. Genet. 1997, 34, 423–425. [Google Scholar] [CrossRef]
- Chaoui, R.; Heling, K.-S.; Zhao, Y.; Sinkovskaya, E.; Abuhamad, A.; Karl, K. Dilated Cavum Septi Pellucidi in Fetuses with Microdeletion 22q11. Prenat. Diagn. 2016, 36, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Pylypjuk, C.L.; Memon, S.F.; Chodirker, B.N. Utility of Measuring Fetal Cavum Septum Pellucidum (CSP) Width During Routine Obstetrical Ultrasound for Improving Diagnosis of 22q11.2 Deletion Syndrome: A Case-Control Study. Appl. Clin. Genet. 2022, 15, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Schindewolf, E.; Crowley, T.B.; McGinn, D.E.; Bailey, A.; Zackai, E.H.; Hopkins, S.E.; Chadehumbe, M.A.; Bilankiuk, L.; Oliver, E.; McDonald-McGinn, D.M.; et al. Dilated Cavum Septum Pellucidum: A Prenatal Soft Marker for Identifying Patients with 22q11.2 Deletion Syndrome. In Proceedings of the Presented at the 12th Biennial International 22q11.2 Scientific Conference, Split, Croatia, 29 June–1 July 2022. [Google Scholar]
- Bohm, L.A.; Zhou, T.C.; Mingo, T.J.; Dugan, S.L.; Patterson, R.J.; Sidman, J.D.; Roby, B.B. Neuroradiographic Findings in 22q11.2 Deletion Syndrome. Am. J. Med. Genet. Part A 2017, 173, 2158–2165. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.E.; Yi, J.J.; Roalf, D.R.; Loevner, L.A.; Ruparel, K.; Whinna, D.; Souders, M.C.; McDonald-McGinn, D.M.; Yodh, E.; Vandekar, S.; et al. Incidental Radiologic Findings in the 22q11.2 Deletion Syndrome. AJNR Am. J. Neuroradiol. 2014, 35, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.S.; Costain, G.; Marshall, C.R. Neuropsychiatric Aspects of 22q11.2 Deletion Syndrome: Considerations in the Prenatal Setting. Prenat. Diagn. 2017, 37, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Kotcher, R.E.; Chait, D.B.; Heckert, J.M.; Crowley, T.B.; Forde, K.A.; Ahuja, N.K.; Mascarenhas, M.R.; Emanuel, B.S.; Zackai, E.H.; McDonald-McGinn, D.M.; et al. Gastrointestinal Features of 22q11.2 Deletion Syndrome Include Chronic Motility Problems from Childhood to Adulthood. J. Pediatr. Gastroenterol. Nutr. 2022, 75, e8. [Google Scholar] [CrossRef]
- Van, L.; Butcher, N.J.; Costain, G.; Ogura, L.; Chow, E.W.C.; Bassett, A.S. Fetal Growth and Gestational Factors as Predictors of Schizophrenia in 22q11.2 Deletion Syndrome. Genet. Med. 2016, 18, 350–355. [Google Scholar] [CrossRef] [Green Version]
- Tramontana, A.; Hartmann, B.; Hafner, E. DiGeorge Syndrome Chromosome Region Deletion and Duplication: Prenatal Genotype-Phenotype Variability in Fetal Ultrasound and MRI. Prenat. Diagn. 2019, 39, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Yerlikaya, G.; Efetürk, T.; Springer, S.; Reischer, T. Prenatal Detection of Right Aortic Arch. Arch. Gynecol. Obs. 2019, 299, 933–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.; Costain, G.; Ogura, L.; Silversides, C.K.; Chow, E.W.C.; Bassett, A.S. Reproductive Health Issues for Adults with a Common Genomic Disorder: 22q11.2 Deletion Syndrome. J. Genet. Couns. 2015, 24, 810–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ESHRE PGT-SR/PGT-A Working Group; Coonen, E.; Rubio, C.; Christopikou, D.; Dimitriadou, E.; Gontar, J.; Goossens, V.; Maurer, M.; Spinella, F.; Vermeulen, N.; et al. ESHRE PGT Consortium Good Practice Recommendations for the Detection of Structural and Numerical Chromosomal Aberrations. Hum. Reprod. Open 2020, 2020, hoaa017. [Google Scholar] [CrossRef]
- Iwarsson, E.; Ahrlund-Richter, L.; Inzunza, J.; Fridström, M.; Rosenlund, B.; Hillensjö, T.; Sjöblom, P.; Nordenskjöld, M.; Blennow, E. Preimplantation Genetic Diagnosis of DiGeorge Syndrome. Mol. Hum. Reprod. 1998, 4, 871–875. [Google Scholar] [CrossRef] [Green Version]
- Shefi, S.; Raviv, G.; Rienstein, S.; Barkai, G.; Aviram-Goldring, A.; Levron, J. Fish Based Preimplantation Genetic Diagnosis to Prevent DiGeorge Syndrome. J. Assist. Reprod. Genet. 2009, 26, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, S.; Crowley, T.B.; Bailey, A.; McGinn, D.E.; Lairson, L.A.; Giunta, V.; Tran, O.; Zackai, E.H.; Emanuel, B.S.; McDonald-McGinn, D.M. Unexpected 22q11.2 Laboratory Results: Parental CNVs, Somatic Mosaicism, and Presumed Germline Mosaicism. In Proceedings of the Presented at the 12th Biennial International 22q11.2 Conference, Split, Croatia, June 28–July 1 2022. [Google Scholar]
- Sandrin-Garcia, P.; Macedo, C.; Martelli, L.; Ramos, E.; Guion-Almeida, M.; Richieri-Costa, A.; Passos, G. Recurrent 22q11.2 Deletion in a Sibship Suggestive of Parental Germline Mosaicism in Velocardiofacial Syndrome. Clin. Genet. 2002, 61, 380–383. [Google Scholar] [CrossRef]
- Vergaelen, E.; Swillen, A.; Van Esch, H.; Claes, S.; Van Goethem, G.; Devriendt, K. 3 Generation Pedigree with Paternal Transmission of the 22q11.2 Deletion Syndrome: Intrafamilial Phenotypic Variability. Eur. J. Med. Genet. 2015, 58, 244–248. [Google Scholar] [CrossRef]
- McGinn, D.E.; Crowley, T.B.; Heung, T.; Tran, O.; Moss, E.; Zackai, E.H.; Emanuel, B.S.; Chow, E.W.C.; Morrow, B.E.; Swillen, A.; et al. Influence of Parent-of-Origin on Intellectual Outcomes in the Chromosome 22q11.2 Deletion Syndrome. Genes 2022, 13, 1800. [Google Scholar] [CrossRef]
- Fiksinski, A.M.; Heung, T.; Corral, M.; Breetvelt, E.J.; Costain, G.; Marshall, C.R.; Kahn, R.S.; Vorstman, J.A.S.; Bassett, A.S. Within-Family Influences on Dimensional Neurobehavioral Traits in a High-Risk Genetic Model. Psychol. Med. 2022, 52, 3184–3192. [Google Scholar] [CrossRef] [PubMed]
- Tomita-Mitchell, A.; Mahnke, D.K.; Larson, J.M.; Ghanta, S.; Feng, Y.; Simpson, P.M.; Broeckel, U.; Duffy, K.; Tweddell, J.S.; Grossman, W.J.; et al. Multiplexed Quantitative Real-Time PCR to Detect 22q11.2 Deletion in Patients with Congenital Heart Disease. Physiol. Genom. 2010, 42A, 52–60. [Google Scholar] [CrossRef]
- Ron, H.; Crowley, T.B.; Liu, Y.; Unolt, M.; Schindewolf, E.; Moldenhauer, J.S.; Rychik, J.; Goldmuntz, E.; Emanuel, B.S.; Ryba, D.; et al. Improved Outcomes in Patients with 22q11.2 Deletion Syndrome and Diagnosis of Interrupted Aortic Arch Prior to Birth Hospital Discharge. Genes 2023, 14, 62. [Google Scholar] [CrossRef]
- Saitta, S.C.; Harris, S.E.; McDonald-McGinn, D.M.; Emanuel, B.S.; Tonnesen, M.K.; Zackai, E.H.; Seitz, S.C.; Driscoll, D.A. Independent de Novo 22q11.2 Deletions in First Cousins with DiGeorge/Velocardiofacial Syndrome. Am. J. Med. Genet. Part A 2004, 124A, 313–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.L.; Crowley, T.B.; McGinn, D.E.; McDougall, C.; Unolt, M.; Lambert, M.P.; Emanuel, B.S.; Zackai, E.H.; McDonald-McGinn, D.M. 22q and Two: 22q11.2 Deletion Syndrome and Coexisting Conditions. Am. J. Med. Genet. A 2018, 176, 2203–2214. [Google Scholar] [CrossRef]
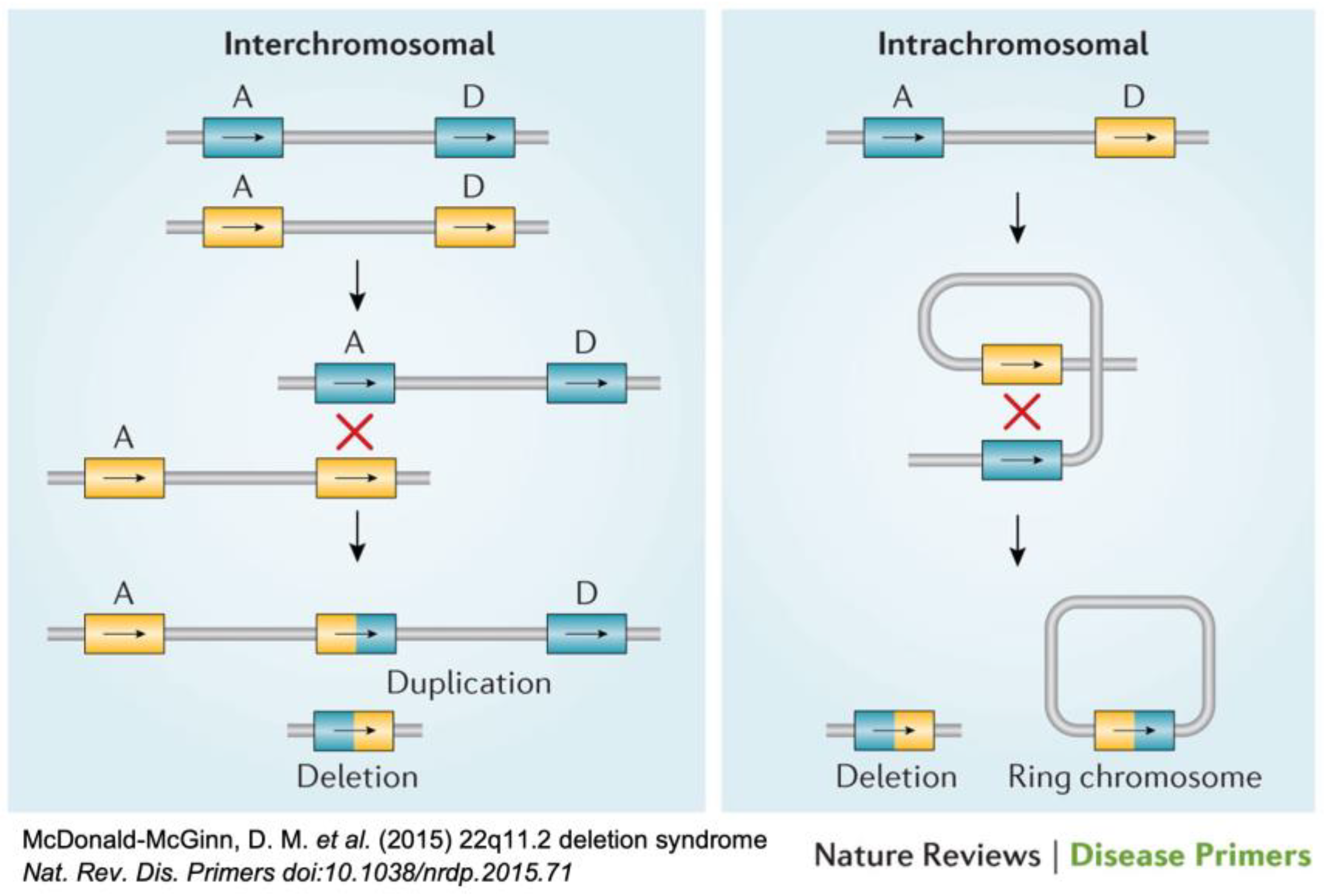




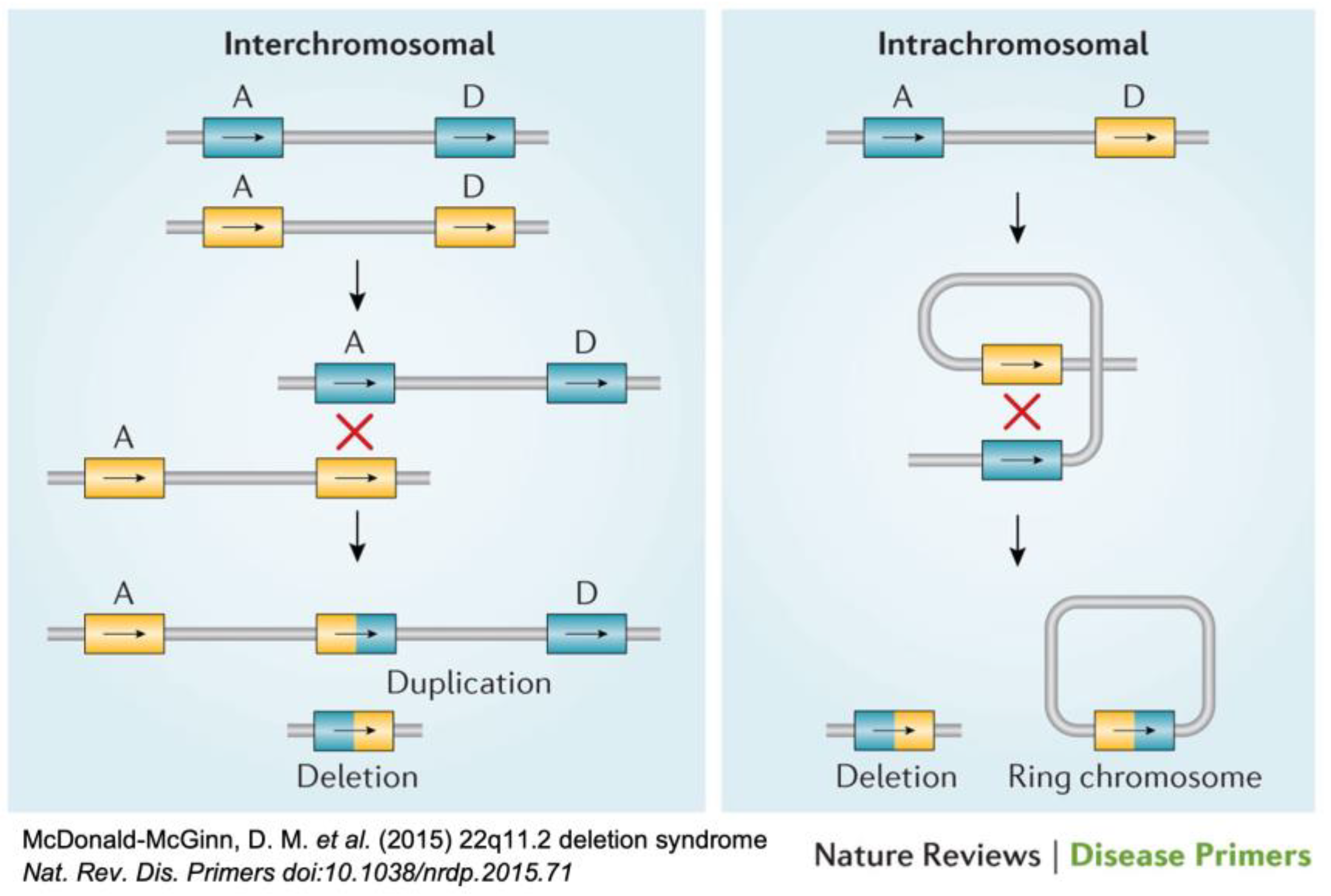{kind=link}
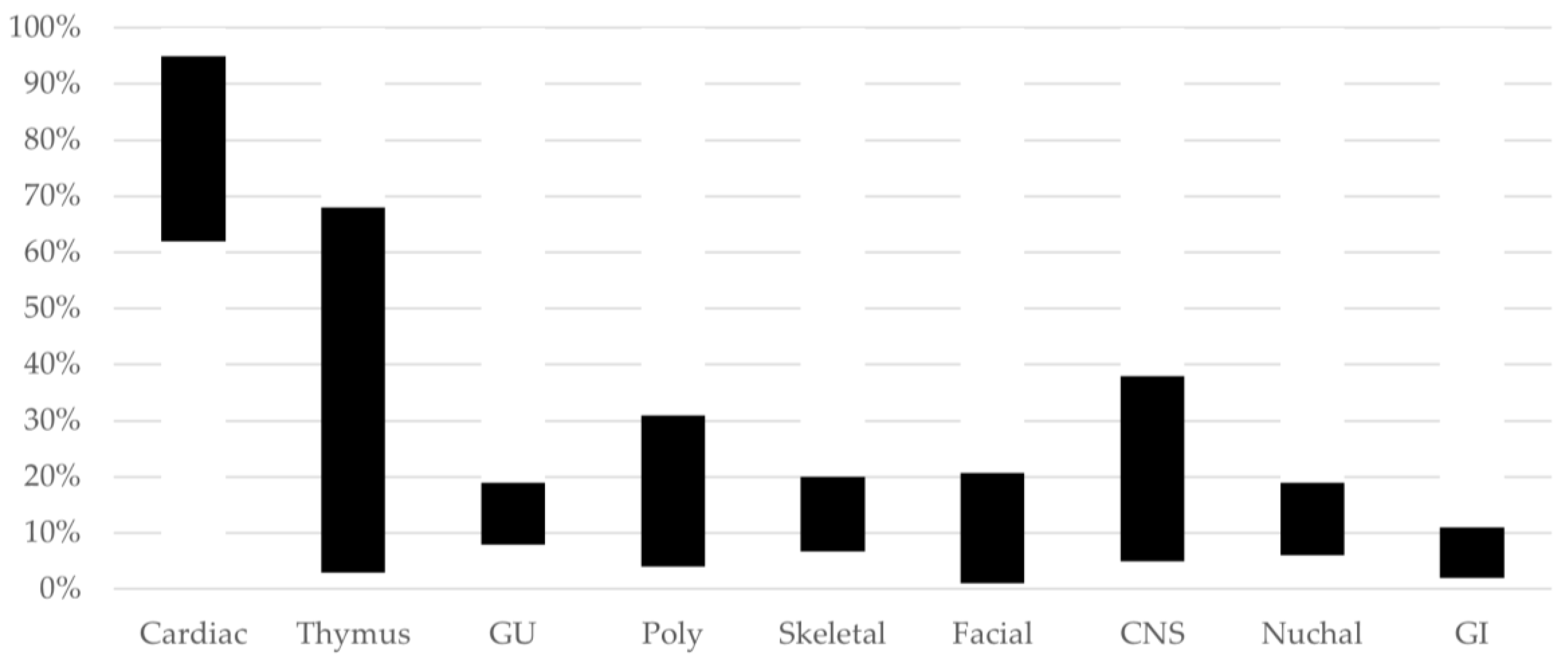
{kind=link}
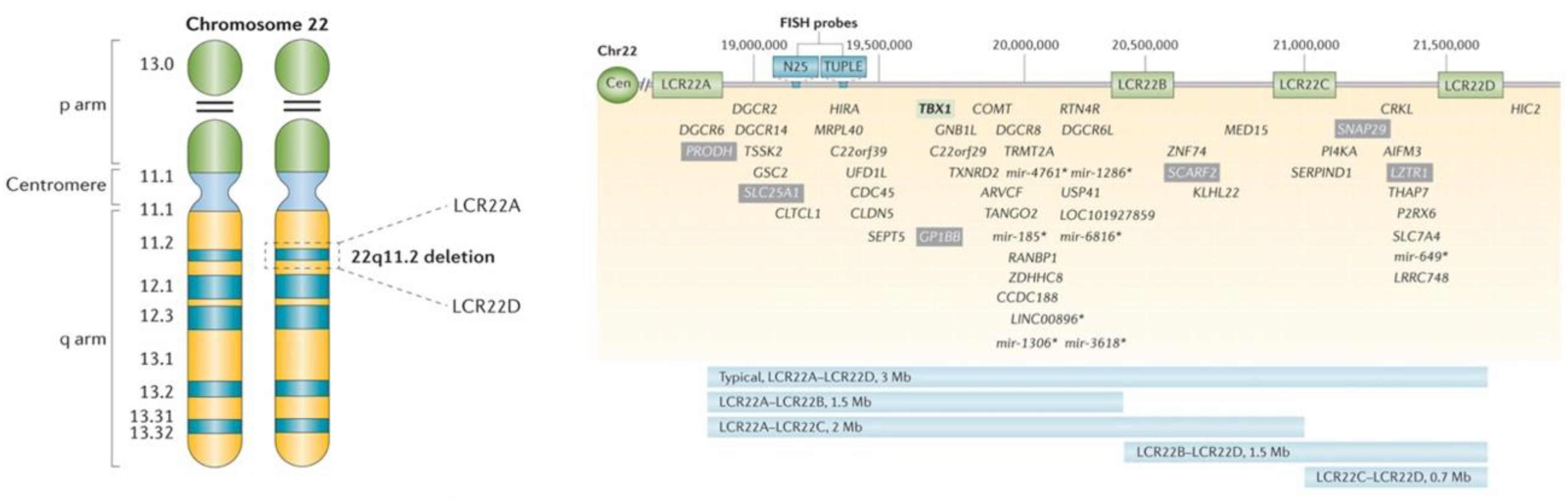
{kind=link}
{kind=link}
{kind=link}
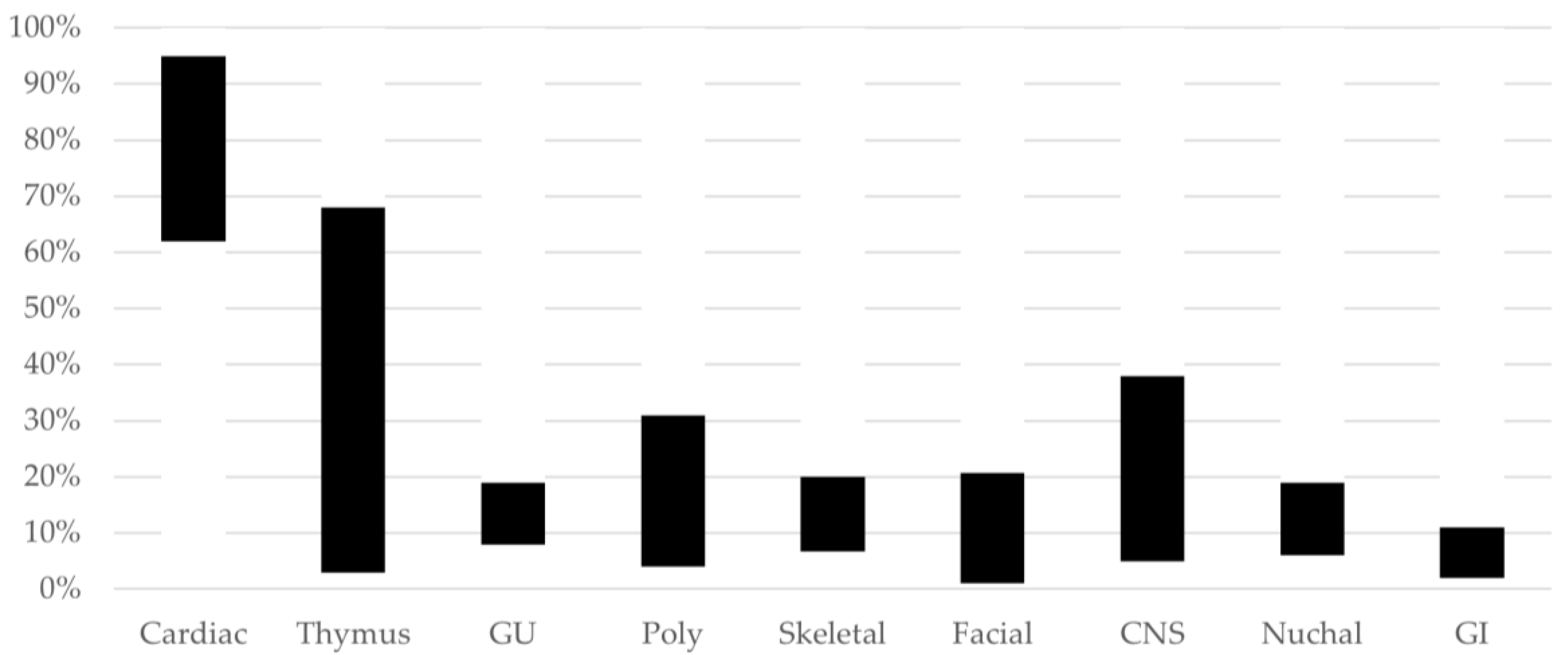
| System Affected | Examples |
|---|---|
| Congenital heart disease (CHD) (64%) [10] (conotruncal anomalies, aortic arch anomalies) | Tetralogy of Fallot (ToF) (18%) [10] Pulmonary atresia ± ventricular septal defect (PA/VSD) (6%) [10] Truncus arteriosus (TA) (4%) [10] Interrupted aortic arch (especially type B) (IAA) (symptomatic once the ductus arteriosus has spontaneously closed, which may be post neonatal discharge) (11%) [10] Conoventricular septal defects (VSD)/muscular VSD (23%) [10] Aortic arch and epiaortic vessel anomalies (AAA), including double aortic arch or right aortic arch (RAA), with/without aberrant subclavian arteries (ASC) ± resulting in a double aortic arch/vascular ring (14%) [10] |
| Palatal abnormalities (67%) [10] | Velopharyngeal insufficiency (most frequently manifesting in infancy as nasal regurgitation) (52%) [10] Submucosal cleft palate ± bifid uvula (21%) [10] Overt cleft palate (6%) [10] Cleft lip and palate (1–2%) [1] |
| Genitourinary anomalies | Renal anomalies (16%) [10] Bilateral or unilateral renal agenesis Multicystic dysplastic kidneys Hyperechoic kidneys Vesicoureteral reflux Inguinal hernia Hypospadias (4%) [10] Cryptorchidism (4%) [10] |
| Skeletal differences | Scoliosis (50%—most frequent onset in adolescence) [10] Cervical spine anomalies (46%—unlikely to be observed prenatally) [10] Talipes [11] Butterfly vertebrae [11] Preaxial and postaxial polydactyly of the hands [1,10] Postaxial polydactyly of the feet [1,10] |
| Immunodeficiency-related | Thymic aplasia or hypoplasia [11] T cell lymphopenia (50%) [10] |
| Endocrinopathies (>50%) [10] | Neonatal hypocalcemia due to hypoparathyroidism ± with neonatal seizures (55%) [10] |
| Otolaryngologic anomalies | Laryngeal web/subglottic stenosis (21%) [10] Trachea-esophageal fistula Esophageal atresia, tracheal atresia |
| Gastrointestinal problems (65%) [10] | Feeding and swallowing difficulties (30%) [10] Umbilical hernia Omphalocele [11] Imperforate anus [1,10] Intestinal malrotation or non-rotation [1,10] Hirschsprung’s disease [1,10] Congenital diaphragmatic hernia (CDH) [1,10] |
| Neurologic manifestations | Hypotonia [1,10] Idiopathic seizures (15%) [10] Neonatal seizures/jitteriness/cyanosis (may be due to hypocalcemia) [10] Microcephaly [1] Polymicrogyria, heterotopias [10] Open operculum [1] Chiari malformation [10] Tethered cord [1] Neural tube defects: myelomeningocele, anencephaly [10,11] |
| Reference | Sensitivity% | Specificity% | Positive Predictive Value (PPV)% | Negative Predictive Value (NPV)% |
|---|---|---|---|---|
| Dar et al., 2022 [26] | 83.3 | 99.8 | 52.6 | 99.9 |
| Lin et al., 2021 [28] | 100 | 99.9 | 53.9 | 99.9 |
| Bevilacqua et al., 2021 [29] | 69.6 * | 100 * | 100 * | 98 * |
| Presenting Prenatal Finding | Total | Isolated | Multiple | Additional Findings |
|---|---|---|---|---|
| Cardiac | 59 | 45 | 14 | |
| ToF | 29 | 25 | 4 | Enlarged nuchal, VSD, AAA + ARSA, Pyelectasis, Hydronephrosis |
| AAA | 2 | 1 | 1 | Hydronephrosis + megaureter |
| ARSA | 2 | - | 2 | Cardiac outflow tracts, VSD |
| IAA | 3 | 3 | - | - |
| Truncus arteriosus | 2 | 1 | 1 | VSD |
| Truncus arteriosus communis | 2 | - | 2 | VSD, RAA |
| Coarctation of aorta | 1 | - | 1 | VSD |
| HLHS | 7 | 6 | 1 | Omphalocele |
| VSD | 11 | 9 | 2 | Micrognathia, Polydactyly |
| Other ultrasound findings | 9 | 7 | 2 | |
| Enlarged nuchal | 2 | 2 | - | - |
| Urinary tract | 3 | 3 | - | Renal diastases, Megacystic |
| CDH | 1 | 1 | - | - |
| Cerebral ventriculomegaly | 1 | - | 1 | Hernia + family history |
| Talipes equinovarus | 1 | 1 | - | - |
| Multiple anomalies | 1 | - | 1 | Not specified |
| Abnormal screening | 11 | 10 | 1 | |
| Serum screening elevated for trisomy | 10 | 9 | 1 | Umbilical hernia |
| NIPS high risk 22q11.2 deletion | 1 | 1 | - | - |
| Parent with 22q11.2 deletion | 2 | 2 | - |
| Genetic diagnosis of 22q11.2 microdeletion in conceptus/embryo/fetus |
|
| Genetic screening of 22q11.2 microdeletion in embryo/fetus |
Noninvasive prenatal genetic screen (NIPS)
|
| When fetus has tested positive for a 22q11.2 microdeletion |
|
| When a parent has a 22q11.2 microdeletion |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blagowidow, N.; Nowakowska, B.; Schindewolf, E.; Grati, F.R.; Putotto, C.; Breckpot, J.; Swillen, A.; Crowley, T.B.; Loo, J.C.Y.; Lairson, L.A.; et al. Prenatal Screening and Diagnostic Considerations for 22q11.2 Microdeletions. Genes 2023, 14, 160. https://doi.org/10.3390/genes14010160
Blagowidow N, Nowakowska B, Schindewolf E, Grati FR, Putotto C, Breckpot J, Swillen A, Crowley TB, Loo JCY, Lairson LA, et al. Prenatal Screening and Diagnostic Considerations for 22q11.2 Microdeletions. Genes. 2023; 14(1):160. https://doi.org/10.3390/genes14010160
Chicago/Turabian StyleBlagowidow, Natalie, Beata Nowakowska, Erica Schindewolf, Francesca Romana Grati, Carolina Putotto, Jeroen Breckpot, Ann Swillen, Terrence Blaine Crowley, Joanne C. Y. Loo, Lauren A. Lairson, and et al. 2023. "Prenatal Screening and Diagnostic Considerations for 22q11.2 Microdeletions" Genes 14, no. 1: 160. https://doi.org/10.3390/genes14010160
APA StyleBlagowidow, N., Nowakowska, B., Schindewolf, E., Grati, F. R., Putotto, C., Breckpot, J., Swillen, A., Crowley, T. B., Loo, J. C. Y., Lairson, L. A., Óskarsdóttir, S., Boot, E., Garcia-Minaur, S., Cristina Digilio, M., Marino, B., Coleman, B., Moldenhauer, J. S., Bassett, A. S., & McDonald-McGinn, D. M. (2023). Prenatal Screening and Diagnostic Considerations for 22q11.2 Microdeletions. Genes, 14(1), 160. https://doi.org/10.3390/genes14010160