
Impact of FecB Mutation on Ovarian DNA Methylome in Small-Tail Han Sheep
Abstract
:1. Introduction
2. Methods
2.1. Experimental Animals
2.2. The Preparation of Sample
2.3. MeDIP-seq Analysis
2.4. MeDIP-seq Sequence Alignments and Data Analysis
2.5. Co-Analysis of Differentially Expressed and Methylated Genes
2.6. Bisulfite Sequencing
2.7. Statistical Analysis
3. Results
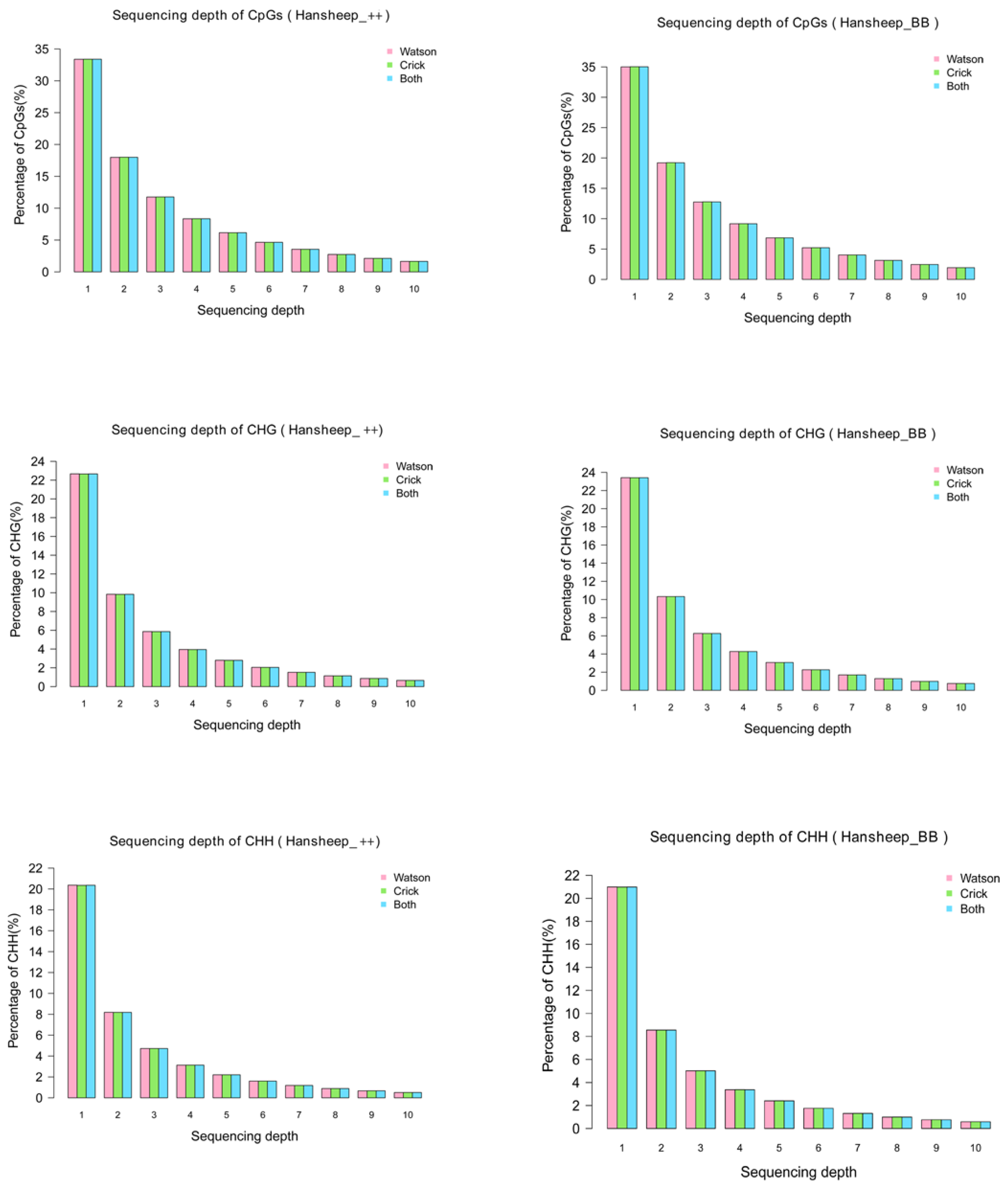
3.1. Global Patterns of DNA Methylation in Han ++ and HanBB Sheep
3.2. DNA Methylation Peaks in Han ++ and HanBB Genome
3.3. Differentially Methylated Genes between Han ++ and Han BB Ewe’s Ovaries
3.4. GO Annotation of Differentially Methylated Genes
3.5. KEGG Pathway Analysis of Differentially Methylated Genes
3.6. MeDIP-seq Data Validation
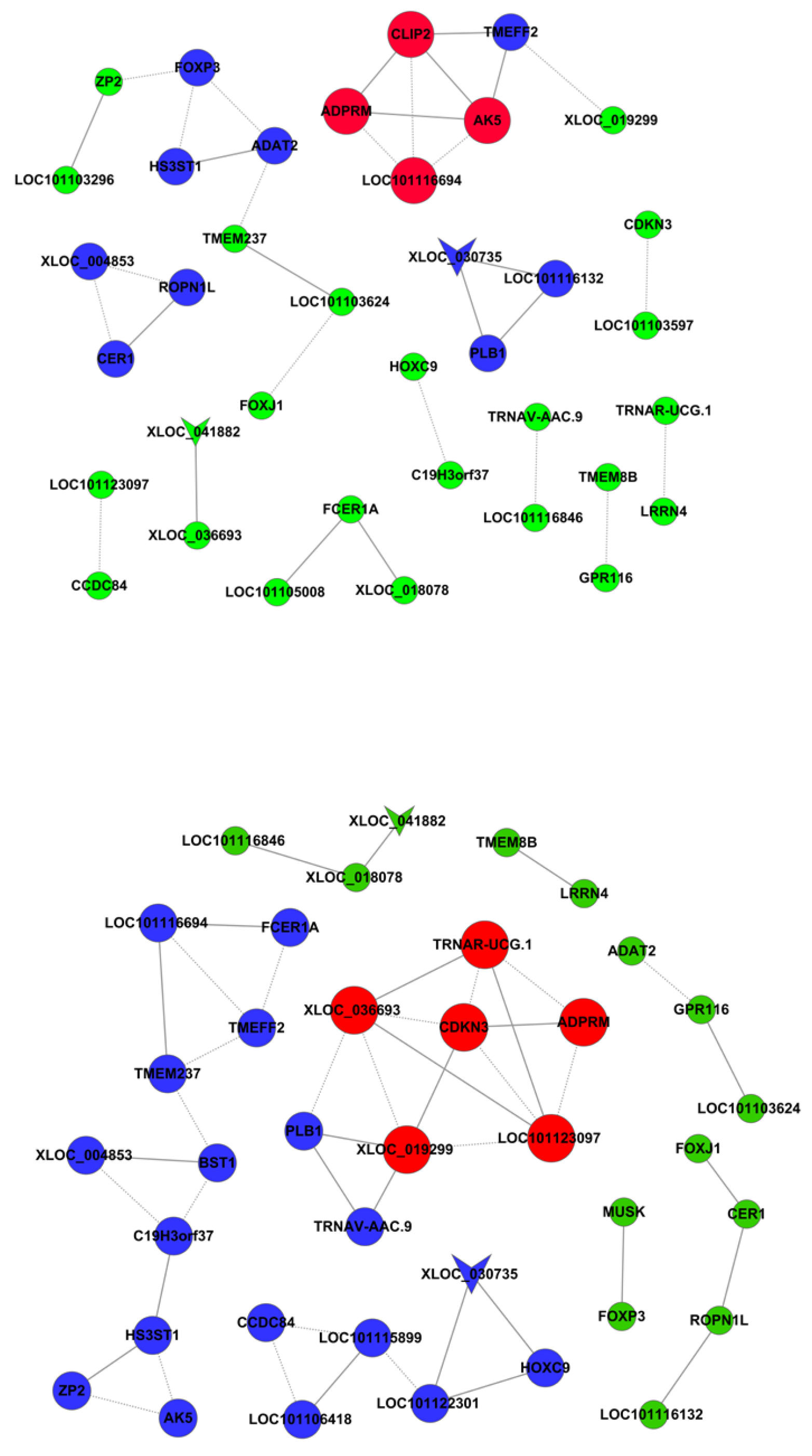
3.7. DNA Methylation and Gene Expression Co-Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hunter, M.G.; Biggs, C.; Faillace, L.S.; Picton, H.M. Current concepts of folliculogenesis in monovular and polyovular farm species. J. Reprod. Fertil. 1992, 45, 21–38. [Google Scholar]
- Fierro, S.; Gil, J.; Vinoles, C.; Olivera-Muzante, J. The use of prostaglandins in controlling estrous cycle of the ewe: A review. Theriogenology 2013, 79, 399–408. [Google Scholar] [CrossRef]
- Davis, G.H. Fecundity genes in sheep. Anim. Reprod. Sci. 2004, 82–83, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Piper, L.R.; Bindon, B.M.; Davis, G.H. The single gene inheritance of the high litter size of the Booroola Merino. In Genetics of Reproduction in Sheep; Land, R.B., Robinson, D.W., Eds.; Butterworths: London, UK, 1985. [Google Scholar]
- Wilson, T.; Wu, X.Y.; Juengel, J.L.; Ross, I.K.; Lumsden, J.M.; Lord, E.A.; Dodds, K.G.; Walling, G.A.; McEwan, J.C.; O’Connell, A.R.; et al. Highly prolific Booroola sheep have a mutation in the intracellular kinase domain of bone morphogenetic protein IB receptor (ALK-6) that is expressed in both oocytes and granulosa cells. Biol. Reprod. 2001, 64, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.J.; MacDougall, C.; MacDougall, C.; Campbell, B.K.; McNeilly, A.S.; Baird, D.T. The Booroola (FecB) phenotype is associated with a mutation in the bone morphogenetic receptor type 1 B (BMPR1B) gene. J. Endocrinol. 2001, 169, R1–R6. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Mishra, A.K.; Kolte, A.P.; Arora, A.L.; Singh, D.; Singh, V.K. Effects of the Booroola (FecB) genotypes on growth performance, ewe’s productivity efficiency and litter size in Garole x Malpura sheep. Anim. Reprod. Sci. 2008, 105, 319–331. [Google Scholar] [CrossRef]
- Gootwine, E.; Rozov, A.; Bor, A.; Reicher, S. Carrying the FecB (Booroola) mutation is associated with lower birth weight and slower post-weaning growth rate for lambs, as well as a lighter mature bodyweight for ewes. Reprod. Fertil. Dev. 2006, 18, 433–437. [Google Scholar] [CrossRef]
- Hua, G.H.; Yang, L.G. A review of research progress of FecB gene in Chinese breeds of sheep. Anim. Reprod. Sci. 2009, 116, 1–9. [Google Scholar] [CrossRef]
- Wang, G.; Mao, X.; George, H.; Zhao, Z.; Zhang, L.; Zeng, Y. DNA tests in Hu sheep and Han sheep (small tail) showed the existence of Booroola (FecB) mutation. J. Nanjing Agric. Univ. 2003, 26, 104–106. [Google Scholar]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Ovarian transcriptomic analysis reveals the alternative splicing events associated with fecundity in different sheep breeds. Anim. Reprod. Sci. 2018, 198, 177–183. [Google Scholar] [CrossRef]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Ovarian proteomic study reveals the possible molecular mechanism for hyperprolificacy of Small Tail Han sheep. Sci. Rep. 2016, 6, 27606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, X.; Qin, Q.L. Genome-wide transcriptome analysis of mRNAs and microRNAs in Dorset and Small Tail Han sheep to explore the regulation of fecundity. Mol. Cell. Endocrinol. 2015, 402, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.L.; Ning, L.I.; Wei, Z.H.; Zhu, X.P.; Liu, H.Y.; Jia, Z.H. Study on FSHR and LHR mRNA Levels of Different BMPRIB Genotypes of Small Tail Han Sheep During the Oestrum. Agric. Sci. China 2007, 6, 94–99. [Google Scholar] [CrossRef]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Co-expression analysis and identification of fecundity-related long non-coding RNAs in sheep ovaries. Sci. Rep. 2016, 6, 39398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Genome-wide analysis of miRNAs in the ovaries of Jining Grey and Laiwu Black goats to explore the regulation of fecundity. Sci. Rep. 2016, 6, 37983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Ovarian transcriptomic study reveals the differential regulation of miRNAs and lncRNAs related to fecundity in different sheep. Sci. Rep. 2016, 6, 35299. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Adrian, B. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar]
- Miao, X.; Luo, Q.; Xie, L.; Zhao, H.; Qin, X. Comparative DNA methylome analysis of estrus ewes reveals the complex regulatory pathways of sheep fecundity. Reprod. Biol. Endocrinol. 2020, 18, 77. [Google Scholar] [CrossRef]
- Quintero-Elisea, J.A.; Macias-Cruz, U.; Alvarez-Valenzuela, F.D.; Correa-Calderon, A.; Gonzalez-Reyna, A.; Lucero-Magana, F.A.; Soto-Navarro, S.A.; Avendano-Reyes, L. The effects of time and dose of pregnant mare serum gonadotropin (PMSG) on reproductive efficiency in hair sheep ewes. Trop. Anim. Health Prod. 2011, 43, 1567–1573. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.K.; Kuroda, M.I. Noncoding RNAs and intranuclear positioning in monoallelic gene expression. Cell 2007, 128, 777–786. [Google Scholar] [CrossRef]
- Dean, W. DNA methylation and demethylation: A pathway to gametogenesis and development. Mol. Reprod. Dev. 2014, 81, 113–125. [Google Scholar] [CrossRef]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [Green Version]
- Ptak, G.; Matsukawa, K.; Palmieri, C.; Della Salda, L.; Scapolo, P.A.; Loi, P. Developmental and functional evidence of nuclear immaturity in prepubertal oocytes. Hum. Reprod. 2006, 21, 2228–2237. [Google Scholar] [CrossRef] [Green Version]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, S.P.; Navratil, A.M.; Xie, J.; Roberson, M.S. GnRH signaling, the gonadotrope and endocrine control of fertility. Front. Neuroendocrinol. 2010, 31, 322–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skorupskaite, K.; George, J.T.; Anderson, R.A. The kisspeptin-GnRH pathway in human reproductive health and disease. Hum. Reprod. Update 2014, 20, 485–500. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.K.; Baird, D.T.; Souza, C.J.; Webb, R. The FecB (Booroola) gene acts at the ovary: In vivo evidence. Reproduction 2003, 126, 101–111. [Google Scholar] [CrossRef]
- Smith, P.; Hudson, N.L.; Corrigan, K.A.; Shaw, L.; Smith, T.; Phillips, D.J.; McNatty, K.P. Effects of the Booroola gene (FecB(B)) on bodymass, testis development and hormone concentrations during fetal life. J. Reprod. Fertil. 1996, 108, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmioglu, N.; Drong, A.W.; Lockstone, H.; Tapmeier, T.; Hellner, K.; Saare, M.; Laisk-Podar, T.; Dew, C.; Tough, E.; Nicholson, G.; et al. Variability of genome-wide DNA methylation and mRNA expression profiles in reproductive and endocrine disease related tissues. Epigenetics 2017, 12, 897–908. [Google Scholar] [CrossRef]
- Rocchi, M.S.; Wattegedera, S.R.; Frew, D.; Entrican, G.; Huntley, J.F.; McNeilly, T.N. Identification of CD4+CD25 high Foxp3+ T cells in ovine peripheral blood. Vet. Immunol. Immunopathol. 2011, 144, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Mitsumoto, Y.; Kyushiki, H.; Kanemoto, N.; Watanabe, A.; Taniguchi, Y.; Nishino, N.; Okamoto, T.; Kondo, M.; Mori, T.; et al. Identification and characterization of TMEFF2, a novel survival factor for hippocampal and mesencephalic neurons. Genomics 2000, 67, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Labeur, M.; Wolfel, B.; Stalla, J.; Stalla, G.K. TMEFF2 is an endogenous inhibitor of the CRH signal transduction pathway. J. Mol. Endocrinol. 2015, 54, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiapekou, E.; Zapanti, E.; Mastorakos, G.; Loutradis, D. Update on the role of ovarian corticotropin-releasing hormone. Ann. N. Y. Acad. Sci. 2010, 1205, 225–229. [Google Scholar] [CrossRef]
- Kiapekou, E.; Zapanti, E.; Voukelatou, D.; Mavreli, T.; Stefanidis, K.; Drakakis, P.; Mastorakos, G.; Loutradis, D. Corticotropin-releasing hormone inhibits in vitro oocyte maturation in mice. Fertil. Steril. 2011, 95, 1497–1499.e1491. [Google Scholar] [CrossRef]
- Lee, S.M.; Park, J.Y.; Kim, D.S. Methylation of TMEFF2 gene in tissue and serum DNA from patients with non-small cell lung cancer. Mol. Cells 2012, 34, 171–176. [Google Scholar] [CrossRef]
- Gerber, A.P.; Keller, W. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science 1999, 286, 1146–1149. [Google Scholar] [CrossRef]
- Molla-Herman, A.; Vallés, A.M.; Ganem-Elbaz, C.; Antoniewski, C.; Huynh, J.-R. tRNA processing defects induce replication stress and Chk2-dependent disruption of piRNA transcription. EMBO J. 2015, 34, 3009–3027. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| gene_id | Forward Primer | Reverse Primer |
|---|---|---|
| PAGE4 | AGTAGGAATAGTGGGTTGTTGT | CACAAACTTAATATATCAATAAACT |
| ADAT2 | GAAGGGATGATGGTTTATTAGGA | CGAACTATCTCCCTCAACATCAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, L.; Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Impact of FecB Mutation on Ovarian DNA Methylome in Small-Tail Han Sheep. Genes 2023, 14, 203. https://doi.org/10.3390/genes14010203
Xie L, Miao X, Luo Q, Zhao H, Qin X. Impact of FecB Mutation on Ovarian DNA Methylome in Small-Tail Han Sheep. Genes. 2023; 14(1):203. https://doi.org/10.3390/genes14010203
Chicago/Turabian StyleXie, Lingli, Xiangyang Miao, Qingmiao Luo, Huijing Zhao, and Xiaoyu Qin. 2023. "Impact of FecB Mutation on Ovarian DNA Methylome in Small-Tail Han Sheep" Genes 14, no. 1: 203. https://doi.org/10.3390/genes14010203