
Metatranscriptomic Analyses Reveal Important Roles of the Gut Microbiome in Primate Dietary Adaptation

 and
and 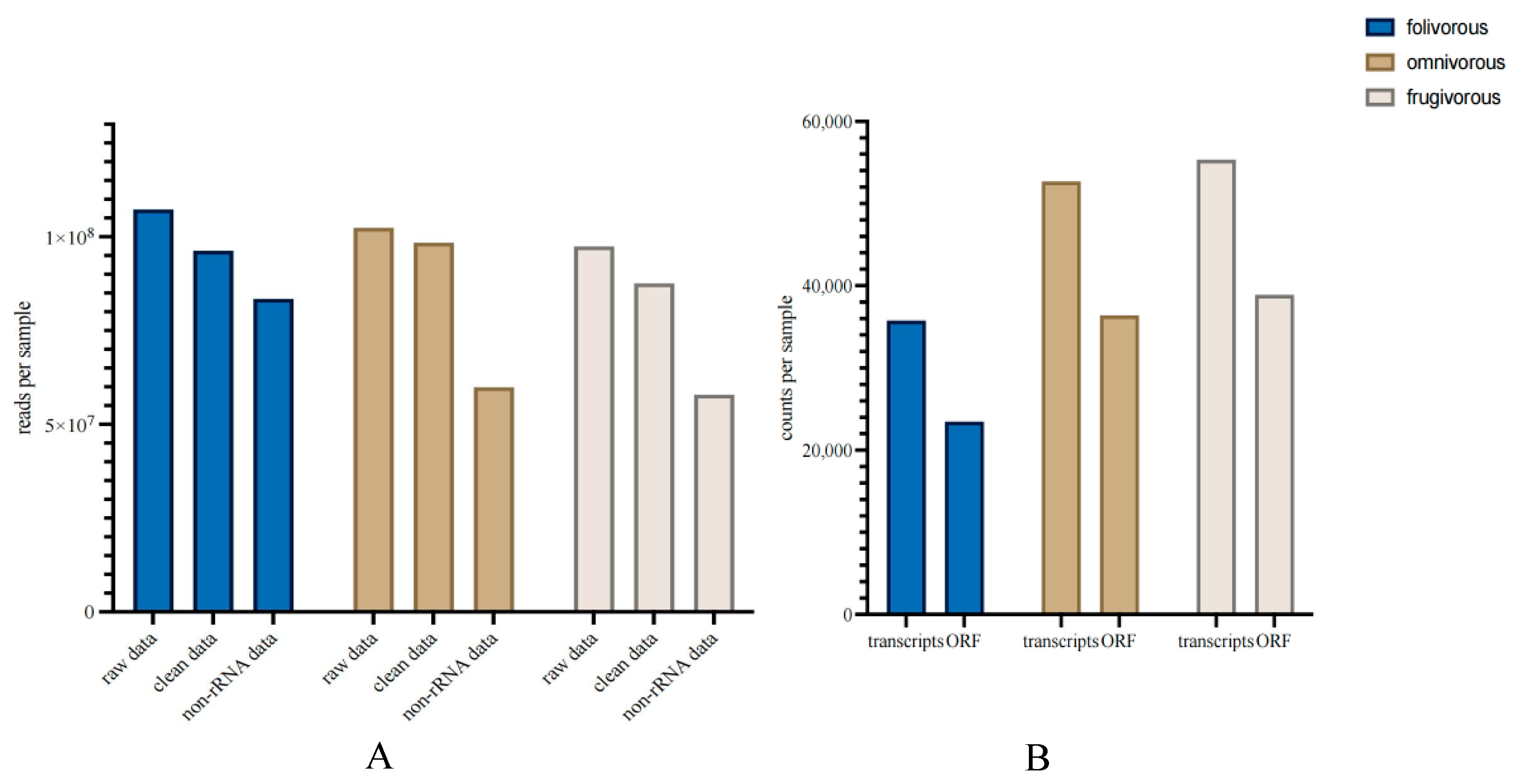{kind=link}
{kind=link}
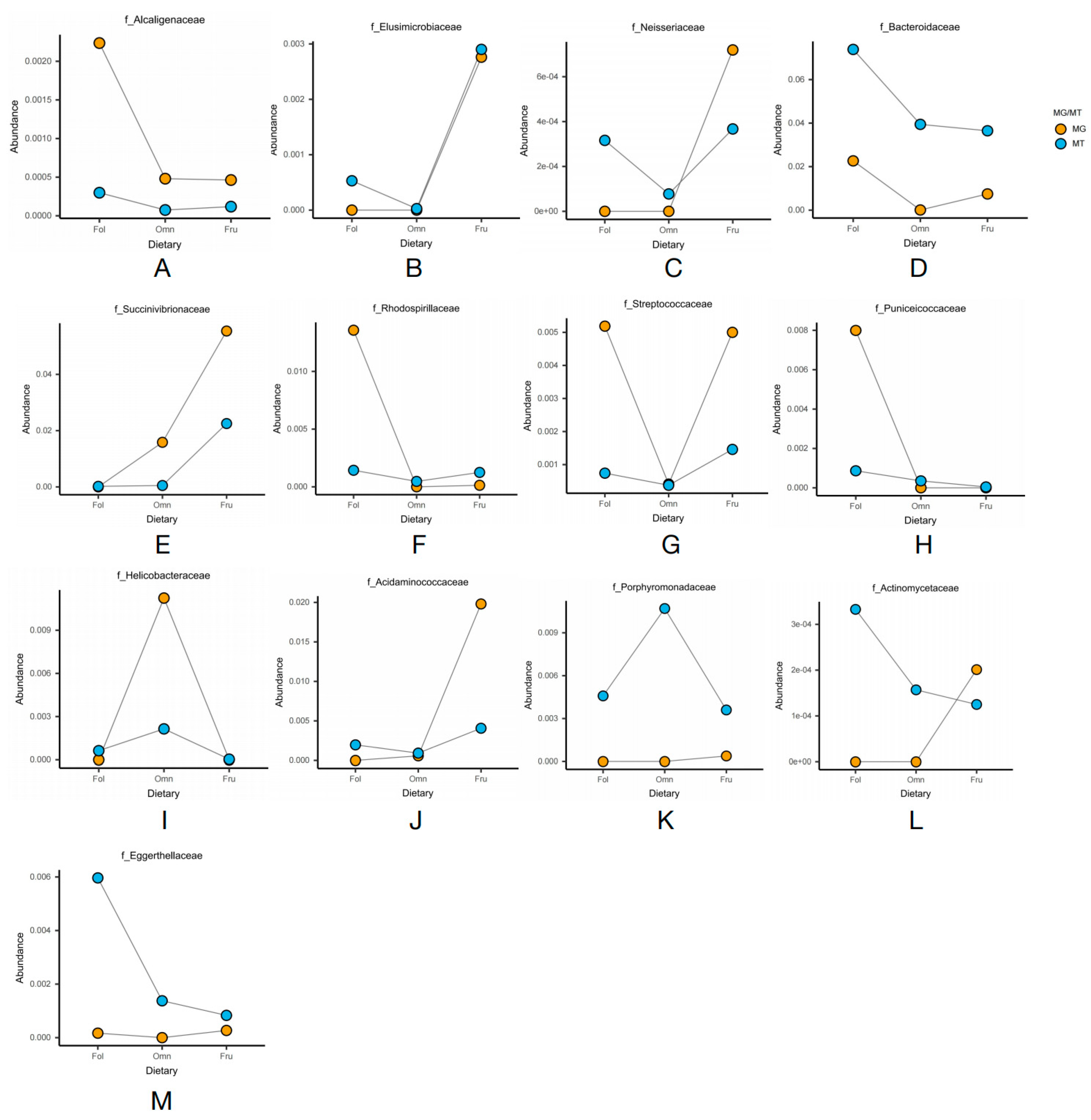{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Extraction, Quality Control and Sequencing
2.3. Construction of Non-Redundant Gene Sets
2.4. Taxonomic Annotation of RNA Non-Redundant Gene Sets
2.5. Functional Annotation of RNA Non-Redundant Gene Sets
2.6. Statistical Analysis
3. Results
3.1. Different Taxa among Three Dietary Primates
3.2. Differences in Gut Microbiome Function among Primates with Three Different Diets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luca, F.; Perry, G.; Di Rienzo, A. Evolutionary adaptations to dietary changes. Annu. Rev. Nutr. 2010, 30, 291. [Google Scholar] [CrossRef]
- Cowlishaw, G.; Dunbar, R.I. Primate conservation biology. Ecology 2001, 82, 1793–1794. [Google Scholar]
- Phillips, K.A.; Bales, K.L.; Capitanio, J.P.; Conley, A.; Czoty, P.W.; ‘t Hart, B.A.; Hopkins, W.D.; Hu, S.L.; Miller, L.A.; Nader, M.A. Why primate models matter. Am. J. Primatol. 2014, 76, 801–827. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R. Incorporating the gut microbiota into models of human and non-human primate ecology and evolution. Am. J. Phys. Anthropol. 2016, 159, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Hanya, G.; Ménard, N.; Qarro, M.; Ibn Tattou, M.; Fuse, M.; Vallet, D.; Yamada, A.; Go, M.; Takafumi, H.; Tsujino, R. Dietary adaptations of temperate primates: Comparisons of Japanese and Barbary macaques. Primates 2011, 52, 187–198. [Google Scholar] [CrossRef]
- Janson, C.H.; Boinski, S. Morphological and behavioral adaptations for foraging in generalist primates: The case of the cebines. Am. J. Phys. Anthropol. 1992, 88, 483–498. [Google Scholar] [CrossRef]
- Borries, C.; Lu, A.; Ossi-Lupo, K.; Larney, E.; Koenig, A. Primate life histories and dietary adaptations: A comparison of Asian colobines and macaques. Am. J. Phys. Anthropol. 2011, 144, 286–299. [Google Scholar] [CrossRef]
- Greene, L.K.; Williams, C.V.; Junge, R.E.; Mahefarisoa, K.L.; Rajaonarivelo, T.; Rakotondrainibe, H.; O’Connell, T.M.; Drea, C.M. A role for gut microbiota in host niche differentiation. ISME J. 2020, 14, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.E. Colobine Monkeys: Their Ecology, Behaviour and Evolution; Cambridge University Press: Cambridge, UK, 1994. [Google Scholar]
- Zhou, X.; Wang, B.; Pan, Q.; Zhang, J.; Kumar, S.; Sun, X.; Liu, Z.; Pan, H.; Lin, Y.; Liu, G. Whole-genome sequencing of the snub-nosed monkey provides insights into folivory and evolutionary history. Nat. Genet. 2014, 46, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Wang, Z.; Zhang, M.; Wang, S.; Xiang, Z.; Pan, H.; Li, M. The Relationship Between Gut Microbiome and Bile Acids in Primates With Diverse Diets. Front. Microbiol. 2022, 13, 899102. [Google Scholar] [CrossRef]
- Lambert, J.E. Primate digestion: Interactions among anatomy, physiology, and feeding ecology. Evol. Anthropol. 1998, 7, 8–20. [Google Scholar] [CrossRef]
- Chivers, D.J.; Davies, A.G.; Oates, J.F. Functional Anatomy of the Gastrointestinal Tract; Cambridge University Press: Cambridge, UK, 1994; pp. 205–227. [Google Scholar]
- Yu, L.; Wang, X.-Y.; Jin, W.; Luan, P.-T.; Ting, N.; Zhang, Y.-P. Adaptive Evolution of Digestive RNASE1 Genes in Leaf-Eating Monkeys Revisited: New Insights from Ten Additional Colobines. Mol. Biol. Evol. 2010, 27, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Fooden, J. Systematic review of the rhesus macaque, Macaca mulatta (Zimmermann, 1780). Fieldiana Zool. 2000, 96, 1–180. [Google Scholar]
- Hill, D.A. Seasonal variation in the feeding behavior and diet of Japanese macaques (Macaca fuscata yakui) in lowland forest of Yakushima. Am. J. Primatol. 1997, 43, 305–322. [Google Scholar] [CrossRef]
- Hon, N.; Behie, A.M.; Rothman, J.M.; Ryan, K.G. Nutritional composition of the diet of the northern yellow-cheeked crested gibbon (Nomascus annamensis) in northeastern Cambodia. Primates 2018, 59, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; de Bruijn, I.; Jack, A.L.; Drynan, K.; van den Berg, A.H.; Thoen, E.; Sandoval-Sierra, V.; Skaar, I.; van West, P.; Dieguez-Uribeondo, J.; et al. Deciphering microbial landscapes of fish eggs to mitigate emerging diseases. ISME J. 2014, 8, 2002–2014. [Google Scholar] [CrossRef]
- Kohl, K.D.; Oakeson, K.F.; Orr, T.J.; Miller, A.W.; Forbey, J.S.; Phillips, C.D.; Dale, C.; Weiss, R.B.; Dearing, M.D. Metagenomic sequencing provides insights into microbial detoxification in the guts of small mammalian herbivores (Neotoma spp.). FEMS Microbiol. Ecol. 2018, 94, fiy184. [Google Scholar] [CrossRef]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012, 6, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Shapira, M. Gut microbiotas and host evolution: Scaling up symbiosis. Trends Ecol. Evol. 2016, 31, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.; Pope, P.B.; Denman, S.E.; McSweeney, C.S. Plant biomass degradation by gut microbiomes: More of the same or something new? Curr. Opin. Biotechnol. 2009, 20, 358–363. [Google Scholar] [CrossRef]
- Roggenbuck, M.; Baerholm Schnell, I.; Blom, N.; Baelum, J.; Bertelsen, M.F.; Sicheritz-Ponten, T.; Sorensen, S.J.; Gilbert, M.T.; Graves, G.R.; Hansen, L.H. The microbiome of New World vultures. Nat. Commun. 2014, 5, 5498. [Google Scholar] [CrossRef] [PubMed]
- Strobel, S.; Roswag, A.; Becker, N.I.; Trenczek, T.E.; Encarnacao, J.A. Insectivorous bats digest chitin in the stomach using acidic mammalian chitinase. PLoS ONE 2013, 8, e72770. [Google Scholar] [CrossRef] [PubMed]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of diet on the gut microbiota: Rethinking intervention duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef] [PubMed]
- Gosalbes, M.J.; Durbán, A.; Pignatelli, M.; Abellan, J.J.; Jimenez-Hernandez, N.; Pérez-Cobas, A.E.; Latorre, A.; Moya, A. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS ONE 2011, 6, e17447. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Xue, X.M.; Kappler, A.; Rosen, B.P.; Meharg, A.A. Linking Genes to Microbial Biogeochemical Cycling: Lessons from Arsenic. Env. Sci. Technol. 2017, 51, 7326–7339. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Gosalbes, M.J.; Friedrichs, A.; Knecht, H.; Artacho, A.; Eismann, K.; Otto, W.; Rojo, D.; Bargiela, R.; von Bergen, M. Gut microbiota disturbance during antibiotic therapy: A multi-omic approach. Gut 2013, 62, 1591–1601. [Google Scholar] [CrossRef]
- Bashiardes, S.; Zilberman-Schapira, G.; Elinav, E. Use of metatranscriptomics in microbiome research. Bioinform. Biol. Insights 2016, 10, BBI.S34610. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- Ismail, W.M.; Ye, Y.; Tang, H. Gene finding in metatranscriptomic sequences. In BMC Bioinformatics; BioMed Central: London, UK, 2014; pp. 1–8. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Salyers, A.; Vercellotti, J.; West, S.; Wilkins, T. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl. Environ. Microbiol. 1977, 33, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Kornman, K.S.; Loesche, W.J. Effects of estradiol and progesterone on Bacteroides melaninogenicus and Bacteroides gingivalis. Infect. Immun. 1982, 35, 256–263. [Google Scholar] [CrossRef]
- Shakya, M.; Lo, C.-C.; Chain, P.S. Advances and challenges in metatranscriptomic analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef]
- de la Garza, A.L.; Romero-Delgado, B.; Martinez-Tamez, A.M.; Cardenas-Tueme, M.; Camacho-Zamora, B.D.; Matta-Yee-Chig, D.; Sanchez-Tapia, M.; Torres, N.; Camacho-Morales, A. Maternal Sweeteners Intake Modulates Gut Microbiota and Exacerbates Learning and Memory Processes in Adult Male Offspring. Front. Pediatr. 2022, 9, 1038. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Liu, Z.; Wang, Y.; Ma, L.; Xu, B. The different dietary sugars modulate the composition of the gut microbiota in honeybee during overwintering. BMC Microbiol. 2020, 20, 1–14. [Google Scholar] [CrossRef]
- Tan, H.; Nie, S. Deciphering diet-gut microbiota-host interplay: Investigations of pectin. Trends Food Sci. Technol. 2020, 106, 171–181. [Google Scholar] [CrossRef]
- Koropatkin, N.M.; Cameron, E.A.; Martens, E.C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 2012, 10, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Riva, A.; Kuzyk, O.; Forsberg, E.; Siuzdak, G.; Pfann, C.; Herbold, C.; Daims, H.; Loy, A.; Warth, B.; Berry, D. A fiber-deprived diet disturbs the fine-scale spatial architecture of the murine colon microbiome. Nat. Commun. 2019, 10, 4366. [Google Scholar] [CrossRef]
- Groussin, M.; Mazel, F.; Sanders, J.G.; Smillie, C.S.; Lavergne, S.; Thuiller, W.; Alm, E.J. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 2017, 8, 14319. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Araujo, M.; Taş, N.; Alcantara-Hernandez, R.J.; Gaona, O.; Schondube, J.E.; Medellin, R.A.; Jansson, J.K.; Falcon, L.I. Phyllostomid bat microbiome composition is associated to host phylogeny and feeding strategies. Front. Microbiol. 2015, 6, 447. [Google Scholar] [CrossRef]
- McKenney, E.A.; Rodrigo, A.; Yoder, A.D. Patterns of gut bacterial colonization in three primate species. PLoS ONE 2015, 10, e0124618. [Google Scholar] [CrossRef]
- Numberger, D.; Herlemann, D.P.; Jürgens, K.; Dehnhardt, G.; Schulz-Vogt, H. Comparative analysis of the fecal bacterial community of five harbor seals (Phoca vitulina). MicrobiologyOpen 2016, 5, 782–792. [Google Scholar] [CrossRef]
- Kartzinel, T.R.; Hsing, J.C.; Musili, P.M.; Brown, B.R.; Pringle, R.M. Covariation of diet and gut microbiome in African megafauna. Proc. Natl. Acad. Sci. USA 2019, 116, 23588–23593. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Conley, M.N.; Roberts, C.; Sharpton, T.J.; Iwaniec, U.T.; Hord, N.G. Increasing dietary nitrate has no effect on cancellous bone loss or fecal microbiome in ovariectomized rats. Mol. Nutr. Food Res. 2017, 61, 1600372. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.O.; Kim, O.-H.; Kim, S.J.; Lee, S.H.; Yun, S.; Lim, S.E.; Yoo, H.J.; Shin, Y.; Lee, S.W. High-fiber diets attenuate emphysema development via modulation of gut microbiota and metabolism. Sci. Rep. 2021, 11, 7008. [Google Scholar] [CrossRef]
- Zhu, L.; Wu, Q.; Dai, J.; Zhang, S.; Wei, F. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 17714–17719. [Google Scholar] [CrossRef]
- Sun, B.; Wang, X.; Bernstein, S.; Huffman, M.A.; Xia, D.-P.; Gu, Z.; Chen, R.; Sheeran, L.K.; Wagner, R.S.; Li, J. Marked variation between winter and spring gut microbiota in free-ranging Tibetan Macaques (Macaca thibetana). Sci. Rep. 2016, 6, 26035. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, K.; Shen, L.; Chen, H.; Hou, F.; Zhou, X.; Zhang, D.; Zhu, X. Microbial community dynamics and assembly follow trajectories of an early-spring diatom bloom in a semienclosed bay. Appl. Environ. Microbiol. 2018, 84, e01000–e01018. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.-W.; Wang, Z.-L.; Shao, Q.; Raubenheimer, D.; Lu, J.-Q. Macronutrient signature of dietary generalism in an ecologically diverse primate in the wild. Behav. Ecol. 2018, 29, 804–813. [Google Scholar] [CrossRef]
- Ali, I.; Koh, Y.-S. High-fat-diet-modulated gut microbiota promotes intestinal carcinogenesis. J. Bacteriol. Virol. 2015, 45, 394–396. [Google Scholar] [CrossRef]
- Trevelline, B.K.; Fontaine, S.S.; Hartup, B.K.; Kohl, K.D. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B 2019, 286, 20182448. [Google Scholar] [CrossRef] [PubMed]
- Mansuy, D. Metabolism of xenobiotics: Beneficial and adverse effects. Biol. Aujourd’hui 2013, 207, 33–37. [Google Scholar] [CrossRef]
- Koppel, N.; Rekdal, V.M.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356, 6344. [Google Scholar] [CrossRef]
- Bianchi, F.; Larsen, N.; de Mello Tieghi, T.; Adorno, M.A.T.; Kot, W.; Saad, S.M.I.; Jespersen, L.; Sivieri, K. Modulation of gut microbiota from obese individuals by in vitro fermentation of citrus pectin in combination with Bifidobacterium longum BB-46. Appl. Microbiol. Biotechnol. 2018, 102, 8827–8840. [Google Scholar] [CrossRef]
- Lutz, H.L.; Jackson, E.W.; Webala, P.W.; Babyesiza, W.S.; Kerbis Peterhans, J.C.; Demos, T.C.; Patterson, B.D.; Gilbert, J.A. Ecology and host identity outweigh evolutionary history in shaping the bat microbiome. Msystems 2019, 4, e00511–e00519. [Google Scholar] [CrossRef]
- Rastelli, M.; Cani, P.D.; Knauf, C. The gut microbiome influences host endocrine functions. Endocr. Rev. 2019, 40, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Berger, P.K.; Plows, J.F.; Alderete, T.L.; Millstein, J.; Fogel, J.; Iablokov, S.N.; Rodionov, D.A.; Osterman, A.L.; Bode, L.; et al. Lactose-reduced infant formula with added corn syrup solids is associated with a distinct gut microbiota in Hispanic infants. Gut Microbes 2020, 12, 1813534. [Google Scholar] [CrossRef] [PubMed]
- France, M.T.; Fu, L.; Rutt, L.; Yang, H.; Humphrys, M.S.; Narina, S.; Gajer, P.M.; Ma, B.; Forney, L.J.; Ravel, J. Insight into the ecology of vaginal bacteria through integrative analyses of metagenomic and metatranscriptomic data. Genome Biol. 2022, 23, 66. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Wang, X.; Wang, Z.; Mao, S.; Zhang, J.; Li, M.; Pan, H. Metatranscriptomic Analyses Reveal Important Roles of the Gut Microbiome in Primate Dietary Adaptation. Genes 2023, 14, 228. https://doi.org/10.3390/genes14010228
Zhang M, Wang X, Wang Z, Mao S, Zhang J, Li M, Pan H. Metatranscriptomic Analyses Reveal Important Roles of the Gut Microbiome in Primate Dietary Adaptation. Genes. 2023; 14(1):228. https://doi.org/10.3390/genes14010228
Chicago/Turabian StyleZhang, Mingyi, Xiaochen Wang, Ziming Wang, Shuxin Mao, Jiali Zhang, Ming Li, and Huijuan Pan. 2023. "Metatranscriptomic Analyses Reveal Important Roles of the Gut Microbiome in Primate Dietary Adaptation" Genes 14, no. 1: 228. https://doi.org/10.3390/genes14010228
APA StyleZhang, M., Wang, X., Wang, Z., Mao, S., Zhang, J., Li, M., & Pan, H. (2023). Metatranscriptomic Analyses Reveal Important Roles of the Gut Microbiome in Primate Dietary Adaptation. Genes, 14(1), 228. https://doi.org/10.3390/genes14010228