Abstract
The overlapping genetic and clinical spectrum in inherited retinal degeneration (IRD) creates challenges for accurate diagnoses. The goal of this work was to determine the genetic diagnosis and clinical features for patients diagnosed with an IRD. After signing informed consent, peripheral blood or saliva was collected from 64 patients diagnosed with an IRD. Genetic testing was performed on each patient in a Clinical Laboratory Improvement Amendments of 1988 (CLIA) certified laboratory. Mutations were verified with Sanger sequencing and segregation analysis when possible. Visual acuity was measured with a traditional Snellen chart and converted to a logarithm of minimal angle of resolution (logMAR). Fundus images of dilated eyes were acquired with the Optos® camera (Dunfermline, UK). Horizontal line scans were obtained with spectral-domain optical coherence tomography (SDOCT; Spectralis, Heidelberg, Germany). Genetic testing combined with segregation analysis resolved molecular and clinical diagnoses for 75% of patients. Ten novel mutations were found and unique genotype phenotype associations were made for the genes RP2 and CEP83. Collective knowledge is thereby expanded of the genetic basis and phenotypic correlation in IRD.
1. Introduction
Non-correctable vision impairments are expected to increase to 13 million people by 2050 [1]. One of the leading causes of these impairments is inherited retinal diseases (IRDs) which has a prevalence of 1 in 2000–3000 [2,3]. IRDs encompass a clinically and genetically heterogeneous group of disorders including retinitis pigmentosa (RP), cone-rod dystrophy (CRD), and various forms of macular dystrophy (MD) such as Stargardt disease (STGD1) and pattern dystrophy. This group of inherited disorders involve progressive degeneration of the retina that leads to severe visual impairment and blindness. The most common IRD, RP is characterized by retinal pigment epithelium (RPE) abnormalities and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptor cells. Patients typically experience a decline to total loss of night vision during adolescence, followed by decreased peripheral vision in young adulthood, and deterioration of the central vision in later life [4]. Dystrophies initially affecting the macula (e.g., MD, STDG1) are distinct from pan-retinal CRD but often have multiple, similar symptoms that can include uncorrectable visual acuity, decreased color perception (dyschromatopsia), and increased sensitivity to light (photophobia).
The overlapping genetic and clinical spectrum in IRD creates challenges for accurate diagnoses. Technological advances in sequencing strategies allow identification of gene mutations for 60–80% of the patients diagnosed with an IRD [5,6,7,8,9,10,11]. IRDs are predominantly Mendelian and monogenic but continue to confound diagnosis due to incomplete penetrance and variable expression. These diseases are typically classified by inheritance type, age of onset, molecular defect, and by distribution of retinal involvement or fundus appearance. A direct link from the molecular and biochemical variations to clinical manifestations contributes to the ideal, unified subclassification system sought by researchers and clinicians alike. This work contributes to realizing this goal.
Genetic diagnosis is a critical step in identifying patients who could be eligible for FDA-approved gene therapy (RPE65-associated IRD-Luxturna [12,13,14]) or for one of the many gene targeted clinical trials (e.g., MERTK, RS1, RPGR-associated IRDs [15,16,17]). An early and accurate diagnosis enables patients to obtain appropriate visual, social, and psychological support that will guide life planning and mitigate disease effects on lifestyle choices. To address these challenges, the objective of this work was to determine the molecular (genetic) basis of IRD for 64 patients paired with clinical findings to advance our understanding of genotype-phenotype associations for IRD.
2. Materials and Methods
Patients were evaluated at Dean McGee Eye Institute (DMEI) at the University of Oklahoma Health Sciences Center (OUHSC) in Oklahoma City, OK. Retrospective data was collected from patients who were seen from 2018 to 2021. All procedures adhered to the Declaration of Helsinki and were approved by institutional ethics review boards. Exclusion criteria included confounding diagnoses such as autoimmune retinopathy, age-related macular degeneration, and history of retinotoxic medications (e.g., pentosan polysulfate or hydroxychloroquine). Demographics in Supplementary Table S1 include identification (ID) number, gender, race or ethnicity, age at diagnosis, age at genetic testing, diagnosis, and inheritance. ID numbers with a letter after the ID number are family members (relation in parenthesis). The ethnicity of the patients in this cohort were White or Caucasian (WC, n = 45, 70.3%), Black or African American (BA, n = 3, 4.7%), Asian (n = 1, 1.6%), Native American (NA, n = 10, 15.6%), unknown or undisclosed (UK, n = 1, 1.6%), and bi-racial (n = 4, 6.3%).
Genetic testing was performed after obtaining informed, written consent from all patients and family members. Peripheral blood or saliva samples of affected and unaffected family members were collected and targeted next generation sequencing (NGS) was performed externally by CLIA certified laboratories (BluePrint Genetics, Invitae, Molecular Vision Laboratory, or Molecular Genetics Laboratory at OUHSC). This certification obliges them to adhere to federal standards. Their testing panels included 351, 330, 1024, and 251 genes, respectively. Molecular Vision Laboratory’s panel included genes for diseases outside the boundaries of IRD. Current IRD gene panels used for NGS are publicly available at Blueprint Genetics, Invitae, and Molecular Vision Laboratory.
Testing performance was provided on individual patient reports. Sequencing was performed using hybridization-based target capture protocols. To summarize the testing information for the aforementioned companies, sequences were mapped to the human reference genome GRCh37/hg19. Genes were excluded from reporting when NGS coverage was suboptimal, i.e., “>90% of the gene’s target nucleotides were not covered at >20x with a mapping quality score of MQ > 20 reads.” Deletions and duplications were identified with proprietary bioinformatics pipelines developed by the individual companies. When deletions, duplications, and pathogenic variants did not meet NGS quality metrics of a given laboratory, orthogonal assays were performed to confirm the results. These assays included, but were not limited to, Pacific Biosciences SMRT sequencing, MLPA, MLPA-seq, Array CGH, and RT-PCR. Each lab designed their own primers and do not provide them on the patient reports. Analytical sensitivity exceeded 99% for variants, including indels less than 15 bp in length for all laboratories. Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines. Pathogenesis of identified variants was determined by the consequences of the change (e.g., amino acid substitution), evolutionary conservation, the number of reference population databases, and mutation databases containing the recorded variant. These included but were not limited to gnomAD, ClinVar, HGMD Professional, and Alamut Visual. Variant calling, methods, and data analysis (or contact information for requests) for individual laboratories can be found at https://blueprintgenetics.com/data-analysis/, https://www.invitae.com/static/data/WhitePaper_Variant-Classification-Method.pdf, https://www.molecularvisionlab.com/company/publications/, or https://genetics.ouhsc.edu/ (accessed on 23 December 2022).
Visual acuity was measured with a traditional Snellen chart in clinic at DMEI and converted to a logarithm of minimal angle of resolution (logMAR). Fundus images of dilated eyes were acquired with the Optos® camera (Optos PLC, Dunfermline, UK). Horizontal line scans were obtained with spectral-domain optical coherence tomography (SDOCT; Spectralis HRA; Heidelberg Engineering, Heidelberg, Germany).
3. Results
3.1. Clinical Assessment
Genetic testing was performed on 64 patients (n = 29 male, 45%; n = 35 female, 55%) diagnosed with an IRD from 53 different families. Clinical diagnosis occurred at an average age of 31.4 ± 15.3 years whereas genetic testing was performed when patients were at the average age of 47.7 ± 16.6 years (Table S1). Clinical diagnoses included RP (n = 32, 50.0%), MD (n = 24, 37.5%), and CRD (n = 8, 12.5%). Refraction and BCVA are shown in Table S1. Ophthalmic evaluations were conducted on 60 patients with IRD (29 RP, 7 CRD, and 23 MD). Overall, patients with RP and CRD had the broadest range of clinical features including pallor of the optic disc, arteriolar narrowing, bone spicules, peripapillary atrophy (PPA), cystoid macular edema (CME), RPE changes in the macula, vitreous syneresis, posterior vitreous detachment (PVD), and/or epiretinal membrane (ERM; Figure 1A). Clinical examination showed typical clinical features (pallor of the optic disc, arteriolar narrowing, and bone spicules) for 17 of 29 individuals diagnosed with RP. Patients with MD primarily displayed RPE changes in the macula (82.6%) and flecks in the posterior pole (73.9%; Figure 1B).
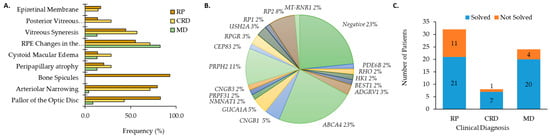
Figure 1.
Clinical characteristics for patients with (A) cone rod dystrophy (CRD) and retinitis pigmentosa (RP) and macular dystrophy (MD). (B) Distribution of genes causative for ocular disease by number of patients (%). (C) The number of genetically solved cases subset by clinical diagnosis.
3.2. Molecular Findings
Genetic results that explain clinical diagnosis were identified for 48 (75%) patients. Pathogenic and likely causative mutations are indicated by the number of patients in this cohort affected by a given gene as shown in Figure 1B. Gene mutations were most frequently (23%) found in the gene ABCA4 (Table 1) followed by PRPH2 (n = 7, 11%; Figure 1B). Five patients from two families had RP2 mutations (8%). Three patients (5%) with autosomal recessive RP (arRP) had CNGB1-related disease. The same GUCA1A mutation (Table 1) was found for three CRD patients (5%) from two different families. Mutations were identified for two individuals for each of the genes ADGRV1, USH2A, and RPGR (3% each). The remaining genes (PDE6B, CNGB3, BEST1, HK1, NMNAT1, PRPF31, RHO, RP1) were associated with a single patient (2%) with IRD (Figure 1B). Ten novel mutations and the previously reported as IRD-causing mutations are provided in Table 1. The unique mutations comprised five missense, one frameshift, three in-frame deletions or duplications, and one promoter mutation (Table 1). Altogether, we were able to provide a genetic diagnosis to 21 RP patients, seven patients with CRD, and 20 patients with MD (Figure 1C).

Table 1.
Patient Genetics.
3.3. Genotype Phenotype Correlations
Pedigree analysis for an RP patient GP006 revealed that his mother (GP006a), maternal grandmother (GP006b), and maternal great uncle (not evaluated, Figure 2A) were also affected. GP006 and GP006a were diagnosed with RP at ages 10 and 15 years, respectively (Table 1). Fundus exam of GP006 showed optic disc pallor OU (oculus uterque or both eyes) arteriolar narrowing, and bone spicules in the peripheral retina (Figure 2B). Hypo-autofluorescence (AF) in the fovea and nasal periphery suggesting RPE atrophy (Figure 2C). SDOCT images revealed ellipsoid zone (EZ) changes and confirmed foveal RPE atrophy OU (Figure 2D). A novel mutation c.515dup (p.Ser172Argfs*2) in the gene RP2 was identified on genetic testing for each of the affected patients (GP006, GP006a, and GP006b). Clinical evaluation for GP006a showed high myopia (Table 1), amblyopia OS (oculus sinister or left eye) and cataract OU. GP006a had typical RP features and RPE changes in the maculae (Figure 2E). Fundus autofluorescence (FAF) imaging displayed nasal hypo-AF regions, with dense hypo-AF areas distributed in the posterior pole (Figure 2F). Additionally, outer nuclear layer (ONL) loss, outer retinal atrophy, and mild vitreomacular adhesion was identified on SDOCT images (Figure 2G). On fundus examination, GP006b showed tilted optic nerves with PPA, arteriolar attenuation, as well as pigmentary and atrophic changes most noticeable in the temporal and inferior periphery (Figure 2H). There were hypo-AF lesions in the inferior periphery and AF striations in the posterior pole OU (Figure 2I). GP006b also displayed significant EZ irregularities including temporal atrophy OU (Figure 2J).
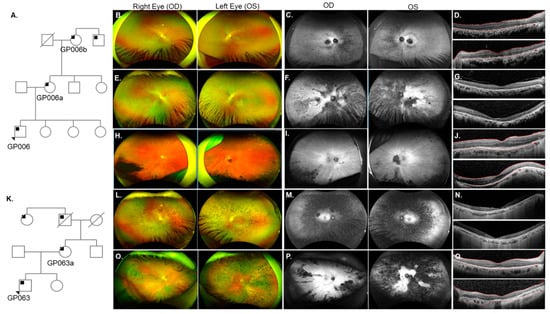
Figure 2.
Phenotype of two families with X-linked RP (XLRP) due to mutations in RP2. (A) Pedigree analysis for patient GP006 showed three generations of RP (black squares). (B) Fundus images for GP006 showed optic disc pallor OU, arteriolar narrowing, and bone spicules in the peripheral retina. (C) Fundus autofluorescence (FAF) revealed hypo-AF in the nasal periphery and fovea. (D) SDOCT showed ellipsoid zone (EZ) changes and retinal pigment epithelium (RPE) atrophy OU. GP006a (E–G) had typical RP features (E) with hypo-AF regions in the nasal retina and throughout the posterior pole (F) and thinning of the outer retina (G). GP006b (H–J) had pallor of the optic disc, chorioretinal atrophy of the far periphery and pigment migration in the inferior retina (H). Radial lines and inferior hypo-AF were noted on FAF (I). SDOCT (J) showed outer retinal atrophy in the temporal macula OU for GP006b. (K) Pedigree analysis for patient GP063 indicated maternally inherited RP with affected family members from three generations. GP063 (L–N) had typical RP features, an atrophic macula, and a loss of EZ band on SDOCT. GP063a (O–Q) had typical RP, multifocal areas of hypo-AF, a loss of EZ temporally OD (upper panel), and complete loss OS (lower panel).
A separate family (patient GP063 and his mother GP063a) also harbored a mutation in RP2 (Figure 2K). GP063 was diagnosed with RP at age 16 (Table S1) but nyctalopia onset occurred at 4-years-of-age. Bilateral cataracts were removed at age 21. GP063 had typical RP features (Figure 2L) and central hypo-AF surrounded by parafoveal hyper-AF (Figure 2M) which was concomitant with photoreceptor loss in the maculae (Figure 2N). The patient’s mother (GP063a) reported nyctalopia at age 16 and was diagnosed with typical RP (Figure 2O) at 30 years-of-age. FAF revealed multifocal areas of hypo-AF corresponding to regions of bone spicule formation on color fundus photos (Figure 2P). Loss of the EZ was seen temporally OD (oculus dexter or right eye) whereas OS had almost complete loss of the photoreceptors (Figure 2Q). Genetic testing for this family revealed a novel RP2 mutation c.413A>C (p.Glu138Ala). A pathogenic mutation has been reported previously at the same codon [87,88,89].
Another interesting case included patient GP037, who was diagnosed with isolate RP at age 30 with nyctalopia onset at 12-years-of-age. Her medical history included hypertension, diabetes, chronic renal insufficiency, gout, and gastric bypass. Fundus images were symmetrical OU showing chorioretinal atrophy, RPE changes in the macula and typical RP features (Figure 3A). FAF showed atrophic midperipheral hypo-AF, PPA, and a bullseye pattern of AF (Figure 3B). SDOCT imaging was notable for outer retinal atrophy with foveal preservation of the EZ (Figure 3C). Four generations of family history revealed no relatives with visual problems (pedigree not shown). Genetic testing reported a pathogenic mutation c.625C>T (p.Arg209*) and a variant of uncertain significance (VUS) c.712A>G (p.Lys238Glu) in the gene CEP83. The VUS Lys238Glu is a missense DNA change encoding a moderately conserved nucleotide present in 8 of 12 total species. ClinVar (2022-02-05, 1005895) classifies this variant with uncertain significance for Nephronophthisis. This information expands the phenotype of IRD for the CEP83 gene which has only been reported in 2 other individuals [58,59] and is discussed below.
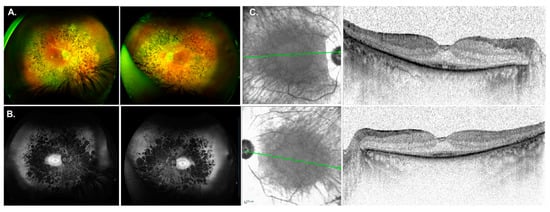
Figure 3.
Phenotype associated with CEP83 mutations. (A) Fundus images for GP037 were showing chorioretinal atrophy, RPE changes in the macula and typical RP features. (B) Fundus autofluorescence imaging showed an atrophic midperiphery and a bullseye pattern of autofluorescence. (C) SDOCT imaging was notable for outer retinal atrophy with foveal preservation of the ellipsoid zone (EZ) line.
4. Discussion
Because IRDs are characterized by genetic and clinical heterogeneity, gene identification and mutation analysis are challenging but important. Continual advancements in sequencing technologies and genetic discoveries have had a substantial impact on the molecular diagnosis of IRD patients [5,6,47,90]. We therefore genetically tested 64 individuals, which identified pathogenic mutations in 18 genes, including ten novel genetic mutations (Table 1). Although patients with RP had the lowest gene detection rate, previous reports using multigene panels, next-generation sequencing, or exome sequencing have had similar findings (50% to 80%) [66,91,92,93,94,95].
A recent study by Sheck et al. [96] found that patients diagnosed with STGD1, MD, or CRD were solved for 91%, 63%, or 87% of patients, respectively. Although we included the STGD1 in the MD group, the diagnostic rates are similar to our results showing that patients with MD and CRD had a 83% and 87% chance of receiving a definitive genetic diagnosis (Figure 1C), respectively. The one patient (GP059) with MD whose test was considered negative, received a partial diagnosis due to a heterozygous pathogenic mutation in ABCA4 c.1749G>C (p.Lys583Asn) [83]. She also harbored a pathogenic mutation in CNGA1 c.1339dup (p.Thr447Asnfs*3). STGD1 MD is inherited in an autosomal recessive manner and no second potentially disease-causing mutation was detected in the ABCA4 gene. It is well known that there are missing alleles in unsolved patients with one pathogenic allele in the ABCA4 gene [97,98].There was a single report (ARVO abstract) suggesting that ABCA4 may cooperate in a digenic fashion to cause IRD [99]. Although a possibility, it is more likely that a second ABCA4 mutation was not detected (intronic or deletion/duplication) or that another yet undiscovered gene is responsible.
Earlier this year, a new gene, CLEC3B, was implicated as a cause of MD [100]. Patients with a CLEC3B mutation had drusen in the posterior pole, nyctalopia, and some decreased rod responses evaluated with full-field electroretinography (ffERG) [100]. This gene was not included in the gene panel for the four MD patients without a molecular diagnosis in our cohort. We therefore used Sanger sequencing to see if these patients had the same mutation. All four patients had the wild type CLEC3B allele (Figure S1). As more information about this gene becomes available, we will consider evaluating the entire coding region of CLEC3B in the unresolved MD cases.
In this research, genotype phenotype relationships were detected for two genes. First, mutations in the gene RP2 were found for two unrelated families. Both RP2 mutations reported here are localized to exon 2, which encodes a cofactor-C homologous domain (CFCHD), confirming a crucial role in RP2 function [101]. The mutation at Ser172 was previously reported as a severe pathogenic frameshift predicted to result in loss of function [36,37]. Jayasundera et al. [36] showed a male patient with this mutation who had significant choriocapillaris atrophy in the midperiphery and the posterior pole without pigment deposition, similar to patient GP006. A different exon 2 mutation (c.413A>C, p.Glu138Ala) in RP2 was found for GP063 and GP063a which affects an evolutionary conserved residue (Glu138) that is a charged, hydrophilic amino acid. Substitution by an uncharged, hydrophobic residue (alanine) is likely to impair CFCHD function, leading to loss or reduced function of the RP2 protein [88]. Multiple lines of computational evidence support a deleterious effect on the gene or gene product (Damaging CADD score of 26.6, likely to interfere with function by Align GVGD Class C65, probably damaging by PolyPhen2, deleterious by SIFT and MutationTaster). This mutation is located in a mutational hot spot and critical functional domain. It is also absent from controls in Exome Sequencing Project, 1000 Genomes Project, and Exome Aggregation Consortium. A different mutation at the same nucleotide (c.413A>G, p.Glu138Gly) was found in two unrelated XLRP families with Italian ancestry [87,88]. Clinical characteristics associated with the p.Glu138Gly mutation included early-onset macular atrophy, progressive BCVA loss, night blindness, and visual field constriction since infancy. Additionally, the patient displayed high refractive myopia, posterior subcapsular cataract, and advanced RP associated with a choroideremia-like fundus [87] similar to other RP2-XLRP patients [36]. Two carrier females (GP006a and GP063a) manifested an RP phenotype, exhibiting atrophic macular changes and poor visual acuity. The carrier female GP006a demonstrated anisometropia (asymmetric refraction) of approximately 2.75 D OS, and her son GP006 was moderately myopic (−3.75 D OU). These refractive errors are in agreement with the myopia association in RP2 retinal disease [36,83,102]. Conversely, GP063 and GP063a were mildly hyperopic in one eye (OD: +0.50 and +0.75, respectively; Table 1) although GP063a had cataract surgery and reported that she was myopic prior to the surgery. There have been no other carrier females with similar mutations described in the RP2-XLRP literature.
To date, RetNet catalogues 340 genes that cause IRDs. However, the gene CEP83 is not included in that database likely owing to its novelty for association with IRD. The CEP83 gene encodes a protein that is essential for ciliogenesis initiation. The CEP83 protein is a core component of the centriole that is found in almost all cell types. Ciliopathies manifest particularly during early childhood or adolescence and can affect almost every organ system [103,104]. Mutations resulting in ciliopathies are often associated with retinal degeneration, cystic kidney disease, fibrocystic liver disease, diabetes mellitus, obesity, skeletal dysplasia, and various abnormalities of the central nervous system [104,105]. GP037 had CEP83 mutations c.625C>T (p.Arg209*) and c.712A>G (p.Lys238Glu). Although CEP83 is primarily associated with nephronophthisis, there have been two publications showing four patients who had autosomal recessive RP (arRP) without nephronophthisis. One of these patients was homozygous for the p.Arg209* mutation with the same clinical features as the patient (GP037) reported here. The biallelic mutations in CEP83 was c.712A>G (p.Lys238Glu). Although pathogenicity has not been proven, we consider the p.Lys238Glu mutation to be disease causing. First, based on the ACMG guidelines, pathogenicity of this mutation is supported by lines of evidence supporting evolutionary conservation (pathogenic supporting argument 3). There is an almost complete absence of the mutation in population databases gnomAD 0.00040084% (1 person) and 0 individuals in ESP, thereby fulfilling a pathogenic moderate argument 2 [106]. Pathogenicity is also supported by in silico modeling (CADD sore 24.1, Align GVGD Class C15, SIFT Deleterious). Moreover, the phenotype suggests pathogenicity (supporting argument 4) of the mutation, as retinal dystrophy has also been reported in another patient with arRP and biallelic mutations in CEP83 (p.Arg209*/p.Arg511Pro) [58] located in the same coiled-coil domain as the p.Lys238Glu mutation harbored by GP057. She received genetic testing in 2017 that revealed a single ABCA4 mutation and a single FAM161A mutation. This was prior to the addition of CEP83 to ophthalmology gene panels. Retesting in 2020 found the two CEP83 mutations in addition to the previously determined pathogenic mutations. Therefore, when a result is incomplete or inconclusive, the patient should be retested after some time because new genes, mutations, and technologies advance rapidly.
These results serve as a basis for future investigation toward understanding genotype phenotype associations and molecular mechanisms underpinning the pathology that leads to vision loss for patients with IRD. Moreover, the knowledge gained from this work contributes to developing treatment for IRD.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes14010074/s1, Figure S1: The c.539 location in CLEC3B associated with macular disorder is wild type in four patients with similar clinical features. Table S1: Gene Panel Lists.
Author Contributions
Conceptualization, L.D.B.; methodology, L.D.B.; software, L.D.B.; validation, L.D.B., C.K.M. and R.G.C.; formal analysis, L.D.B., C.K.M. and R.G.C.; investigation, L.D.B.; resources, L.D.B.; data curation, B.R., J.R., N.B., R.W.Y. and A.R.; writing—original draft preparation, J.L., C.K.M. and L.D.B.; writing—review and editing, J.L., N.B., J.R., R.W.Y., A.R., R.G.C. and R.K.; visualization, L.D.B.; supervision, R.K., R.G.C. and L.D.B.; project administration, L.D.B.; funding acquisition, L.D.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Eye Institute at the National Institutes of Health (NIH), R00EYE027460 (L.D.B). This research was also supported by the Oklahoma Shared Center for Translational Research (U54GM104938, PI James) and the Presbyterian Health Foundation (PHF). This work was supported in part by NIH grant P30EY027125 (NIH CORE grant), and an unrestricted grant to the Dean A. McGee Eye Institute from Research to Prevent Blindness Inc. (http://www.rpbusa.org). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of the University of Oklahoma Health Sciences Center (protocol code 9726 and 11-28-2018).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
NGS panel statistics available upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chan, T.; Friedman, D.S.; Bradley, C.; Massof, R. Estimates of incidence and prevalence of visual impairment, low vision, and blindness in the united states. JAMA Ophthalmol. 2018, 136, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Bessant, D.A.; Ali, R.R.; Bhattacharya, S.S. Molecular genetics and prospects for therapy of the inherited retinal dystrophies. Curr. Opin. Genet. Dev. 2001, 11, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Fahim, A.T.; Daiger, S.P.; Weleber, R.G. Nonsyndromic retinitis pigmentosa overview. In Genereviews((r)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreño, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 2021, 11, 1526. [Google Scholar] [CrossRef]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular Epidemiology in 591 Italian Probands with Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Investig. Opthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef]
- Motta, F.; Martin, R.P.; Filippelli-Silva, R.; Salles, M.V.; Sallum, J.M.F. Relative frequency of inherited retinal dystrophies in Brazil. Sci. Rep. 2018, 8, 15939. [Google Scholar] [CrossRef]
- Whelan, L.; Dockery, A.; Wynne, N.; Zhu, J.; Stephenson, K.; Silvestri, G.; Turner, J.; O’Byrne, J.J.; Carrigan, M.; Humphries, P.; et al. Findings from a Genotyping Study of over 1000 People with Inherited Retinal Disorders in Ireland. Genes 2020, 11, 105. [Google Scholar] [CrossRef]
- Sharon, D.; Ben-Yosef, T.; Cohen, N.G.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2020, 41, 140–149. [Google Scholar] [CrossRef]
- Jacobson, S.G. Gene Therapy for Leber Congenital Amaurosis Caused by RPE65 Mutations. JAMA Ophthalmol. 2012, 130, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; High, K.A.; Auricchio, A.; Wright, J.F.; Pierce, E.A.; Testa, F.; Mingozzi, F.; Bennicelli, J.L.; Ying, G.S.; Rossi, S.; et al. Age-dependent effects of rpe65 gene therapy for leber’s congenital amaurosis: A phase 1 dose-escalation trial. Lancet 2009, 374, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Russell, S.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Drack, A.V.; Simonelli, F.; Leroy, B.P.; Reape, K.Z.; High, K.A.; et al. Durability of Voretigene Neparvovec for Biallelic RPE65-Mediated Inherited Retinal Disease. Ophthalmology 2021, 128, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, N.G.; Abboud, E.B.; Nowilaty, S.R.; Alkuraya, H.; Alhommadi, A.; Cai, H.; Hou, R.; Deng, W.-T.; Boye, S.L.; Almaghamsi, A.; et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: Results of a phase I trial. Hum. Genet. 2016, 135, 327–343. [Google Scholar] [CrossRef]
- Cehajic-Kapetanovic, J.; Xue, K.; de la Camara, C.M.-F.; Nanda, A.; Davies, A.; Wood, L.J.; Salvetti, A.P.; Fischer, M.D.; Aylward, J.W.; Barnard, A.R.; et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 2020, 26, 354–359. [Google Scholar] [CrossRef]
- Parker, M.A.; Erker, L.R.; Audo, I.; Choi, D.; Mohand-Said, S.; Sestakauskas, K.; Benoit, P.; Appelqvist, T.; Krahmer, M.; Ségaut-Prévost, C.; et al. Three-Year Safety Results of SAR422459 (EIAV-ABCA4) Gene Therapy in Patients With ABCA4-Associated Stargardt Disease: An Open-Label Dose-Escalation Phase I/IIa Clinical Trial, Cohorts 1-5. Am. J. Ophthalmol. 2022, 240, 285–301. [Google Scholar] [CrossRef]
- Jiang, L.; Wheaton, D.; Bereta, G.; Zhang, K.; Palczewski, K.; Birch, D.G.; Baehr, W. A novel GCAP1(N104K) mutation in EF-hand 3 (EF3) linked to autosomal dominant cone dystrophy. Vis. Res. 2008, 48, 2425–2432. [Google Scholar] [CrossRef][Green Version]
- Wawrocka, A.; Skorczyk-Werner, A.; Wicher, K.; Niedziela, Z.; Ploski, R.; Rydzanicz, M.; Sykulski, M.; Kociecki, J.; Weisschuh, N.; Kohl, S.; et al. Novel variants identified with next-generation sequencing in Polish patients with cone-rod dystrophy. Mol. Vis. 2018, 24, 326–339. [Google Scholar]
- McLaughlin, M.E.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. Recessive mutations in the gene encoding the β–subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat. Genet. 1993, 4, 130–134. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Mittal, R.; Bencie, N.; Parrish, J.M.; Liu, G.; Mittal, J.; Yan, D.; Liu, X.Z. An Update on Phosphodiesterase Mutations Underlying Genetic Etiology of Hearing Loss and Retinitis Pigmentosa. Front. Genet. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.; Duncan, J.L.; Maranhao, B.; Kozak, I.; Branham, K.; Gabriel, L.; Lin, J.H.; Barteselli, G.; Navani, M.; Suk, J.; et al. Genetic analysis of 10 pedigrees with inherited retinal degeneration by exome sequencing and phenotype-genotype association. Physiol. Genom. 2017, 49, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Nash, B.; Symes, R.; Goel, H.; Dinger, M.E.; Bennetts, B.; Grigg, J.R.; Jamieson, R.V. NMNAT1 variants cause cone and cone-rod dystrophy. Eur. J. Hum. Genet. 2018, 26, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Eblimit, A.; Zaneveld, S.A.; Liu, W.; Thomas, K.; Wang, K.; Li, Y.; Mardon, G.; Chen, R. NMNAT1 E257K variant, associated with Leber Congenital Amaurosis (LCA9), causes a mild retinal degeneration phenotype. Exp. Eye Res. 2018, 173, 32–43. [Google Scholar] [CrossRef]
- Chiang, P.-W.; Wang, J.; Chen, Y.; Fu, Q.; Zhong, J.; Chen, Y.; Yi, X.; Wu, R.; Gan, H.; Shi, Y.; et al. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat. Genet. 2012, 44, 972–974. [Google Scholar] [CrossRef]
- Sears, J.E.; Aaberg, T.A.; Daiger, S.P.; Moshfeghi, D.M. Splice site mutation in the peripherin/RDS gene associated with pattern dystrophy of the retina. Am. J. Ophthalmol. 2001, 132, 693–699. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Lewis, R.A.; Garcia, C.A.; Ruiz, R.S.; Blanton, S.H.; Northrup, H.; et al. Prevalence of Disease-Causing Mutations in Families with Autosomal Dominant Retinitis Pigmentosa: A Screen of Known Genes in 200 Families. Investig. Opthalmol. Vis. Sci. 2006, 47, 3052–3064. [Google Scholar] [CrossRef]
- Zampaglione, E.; Kinde, B.; Place, E.M.; Navarro-Gomez, D.; Maher, M.; Jamshidi, F.; Nassiri, S.; Mazzone, J.A.; Finn, C.; Schlegel, D.; et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Anesth. Analg. 2020, 22, 1079–1087. [Google Scholar] [CrossRef]
- Shankar, S.P.; Birch, D.G.; Ruiz, R.S.; Hughbanks-Wheaton, D.K.; Sullivan, L.S.; Bowne, S.J.; Stone, E.M.; Daiger, S.P. Founder Effect of a c.828+3A>T Splice Site Mutation in Peripherin 2 (PRPH2) Causing Autosomal Dominant Retinal Dystrophies. JAMA Ophthalmol. 2015, 133, 511–517. [Google Scholar] [CrossRef]
- Reeves, M.J.; Goetz, K.E.; Guan, B.; Ullah, E.; Blain, D.; Zein, W.M.; Tumminia, S.J.; Hufnagel, R.B. Genotype–phenotype associations in a large PRPH2 -related retinopathy cohort. Hum. Mutat. 2020, 41, 1528–1539. [Google Scholar] [CrossRef]
- Shankar, S.P.; Hughbanks-Wheaton, D.K.; Birch, D.G.; Sullivan, L.S.; Conneely, K.N.; Bowne, S.J.; Stone, E.M.; Daiger, S.P. Autosomal Dominant Retinal Dystrophies Caused by a Founder Splice Site Mutation, c.828+3A>T, in PRPH2 and Protein Haplotypes in trans as Modifiers. Investig. Opthalmol. Vis. Sci. 2016, 57, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Mauro-Herrera, M.; Chiang, J.; Radojevic, B.; Bennett, L. Functional Evaluation of Splicing for Variants of Uncertain Significance in Patients with Inherited Retinal Diseases. Genes 2021, 12, 993. [Google Scholar] [CrossRef] [PubMed]
- Radojevic, B.; Jones, K.; Klein, M.; Mauro-Herrera, M.; Kingsley, R.; Birch, D.G.; Bennett, L.D. Variable expressivity in patients with autosomal recessive retinitis pigmentosa associated with the gene CNGB1. Ophthalmic Genet. 2021, 42, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Schindler, E.I.; Nylen, E.L.; Ko, A.C.; Affatigato, L.M.; Heggen, A.C.; Wang, K.; Sheffield, V.C.; Stone, E.M. Deducing the pathogenic contribution of recessive ABCA4 alleles in an outbred population. Hum. Mol. Genet. 2010, 19, 3693–3701. [Google Scholar] [CrossRef]
- Jayasundera, K.; Branham, K.; Othman, M.; Rhoades, W.R.; Karoukis, A.J.; Khanna, H.; Swaroop, A.; Heckenlively, J.R. RP2 Phenotype and Pathogenetic Correlations in X-Linked Retinitis Pigmentosa. JAMA Ophthalmol. 2010, 128, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Breuer, D.K.; Yashar, B.M.; Filippova, E.; Hiriyanna, S.; Lyons, R.H.; Mears, A.J.; Asaye, B.; Acar, C.; Vervoort, R.; Wright, A.F.; et al. A Comprehensive Mutation Analysis of RP2 and RPGR in a North American Cohort of Families with X-Linked Retinitis Pigmentosa. Am. J. Hum. Genet. 2002, 70, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Renner, A.B.; Fiebig, B.S.; Weber, B.H.; Wissinger, B.; Andreasson, S.; Gal, A.; Cropp, E.; Kohl, S.; Kellner, U. Phenotypic Variability and Long-term Follow-up of Patients with Known and Novel PRPH2/RDS Gene Mutations. Am. J. Ophthalmol. 2009, 147, 518–530.e1. [Google Scholar] [CrossRef]
- Kohl, S.; Baumann, B.; Broghammer, M.; Jägle, H.; Sieving, P.; Kellner, U.; Spegal, R.; Anastasi, M.; Zrenner, E.; Sharpe, L.T.; et al. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum. Mol. Genet. 2000, 9, 2107–2116. [Google Scholar] [CrossRef]
- Eksandh, L.; Kohl, S.; Wissinger, B. Clinical features of achromatopsia in Swedish patients with defined genotypes. Ophthalmic Genet. 2002, 23, 109–120. [Google Scholar] [CrossRef]
- Johnson, S.; Michaelides, M.; Aligianis, I.A.; Ainsworth, J.R.; Mollon, J.D.; Maher, E.R.; Moore, A.T.; Hunt, D.M. Achromatopsia caused by novel mutations in both CNGA3 and CNGB3. J. Med. Genet. 2004, 41, e20. [Google Scholar] [CrossRef]
- Kohl, S.; Varsanyi, B.; Antunes, G.A.; Baumann, B.; Hoyng, C.B.; Jägle, H.; Rosenberg, T.; Kellner, U.; Lorenz, B.; Salati, R.; et al. CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur. J. Hum. Genet. 2005, 13, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, K.M.; Sandberg, M.A.; Gorji, N.; Berson, E.L.; Dryja, T.P. Cone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseases. Hum. Mutat. 2005, 25, 248–258. [Google Scholar] [CrossRef]
- Wiszniewski, W.; Lewis, R.A.; Lupski, J.R. Achromatopsia: The CNGB3 p.T383fsX mutation results from a founder effect and is responsible for the visual phenotype in the original report of uniparental disomy 14. Qual. Life Res. 2007, 121, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.K.; Van Cauwenbergh, C.; Rother, C.; Baumann, B.; Reuter, P.; De Baere, E.; Wissinger, B.; Kohl, S.; ACHM Study Group. CNGB3 mutation spectrum including copy number variations in 552 achromatopsia patients. Hum. Mutat. 2017, 38, 1579–1591. [Google Scholar] [CrossRef]
- Kinnick, T.R.; Mullins, R.F.; Dev, S.; Leys, M.; Mackey, D.A.; Kay, C.N.; Lam, B.L.; Fishman, G.; Traboulsi, E.; Iezzi, R.; et al. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina 2011, 31, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Jespersgaard, C.; Fang, M.; Bertelsen, M.; Dang, X.; Jensen, H.; Chen, Y.; Bech, N.; Dai, L.; Rosenberg, T.; Zhang, J.; et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci. Rep. 2019, 9, 1219. [Google Scholar] [CrossRef]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Bader, I.; Brandau, O.; Achatz, H.; Apfelstedt-Sylla, E.; Hergersberg, M.; Lorenz, B.; Wissinger, B.; Wittwer, B.; Rudolph, G.; Meindl, A.; et al. X-linked Retinitis Pigmentosa: RPGR Mutations in Most Families with Definite X Linkage and Clustering of Mutations in a Short Sequence Stretch of Exon ORF15. Investig. Opthalmol. Vis. Sci. 2003, 44, 1458–1463. [Google Scholar] [CrossRef]
- Lewis, R.A.; Shroyer, N.F.; Singh, N.; Allikmets, R.; Hutchinson, A.; Li, Y.; Lupski, J.R.; Leppert, M.; Dean, M. Genotype/Phenotype Analysis of a Photoreceptor-Specific ATP-Binding Cassette Transporter Gene, ABCR, in Stargardt Disease. Am. J. Hum. Genet. 1999, 64, 422–434. [Google Scholar] [CrossRef]
- Klevering, B.J.; Yzer, S.; Rohrschneider, K.; Zonneveld, M.; Allikmets, R.; Born, L.I.V.D.; Maugeri, A.; Hoyng, C.B.; Cremers, F.P. Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone–rod dystrophy and retinitis pigmentosa. Eur. J. Hum. Genet. 2004, 12, 1024–1032. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lee, W.; De Carvalho, J.R.L.; Chang, S.; Tsang, S.H.; Allikmets, R.; Sparrow, J.R. Multi-platform imaging in ABCA4-Associated Disease. Sci. Rep. 2019, 9, 6436. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Zernant, J.; Chana, R.K.; Wright, G.A.; Tsunoda, K.; Ozawa, Y.; Tsubota, K.; Webster, A.R.; Moore, A.T.; Allikmets, R.; et al. ABCA4 Gene Screening by Next-Generation Sequencing in a British Cohort. Investig. Opthalmol. Vis. Sci. 2013, 54, 6662–6674. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Héon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- McGee, T.L.; Seyedahmadi, B.J.; Sweeney, M.O.; Dryja, T.P.; Berson, E.L. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J. Med Genet. 2010, 47, 499–506. [Google Scholar] [CrossRef]
- Dreyer, B.; Tranebjærg, L.; Rosenberg, T.; Weston, M.D.; Kimberling, W.J.; Nilssen, O. Identification of novel USH2A mutations: Implications for the structure of USH2A protein. Eur. J. Hum. Genet. 2000, 8, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Failler, M.; Gee, H.Y.; Krug, P.; Joo, K.; Halbritter, J.; Belkacem, L.; Filhol, E.; Porath, J.D.; Braun, D.A.; Schueler, M.; et al. Mutations of CEP83 Cause Infantile Nephronophthisis and Intellectual Disability. Am. J. Hum. Genet. 2014, 94, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Veldman, B.C.F.; Kuper, W.F.E.; Lilien, M.; Schuurs-Hoeijmakers, J.H.M.; Marcelis, C.; Phan, M.; Hettinga, Y.; Talsma, H.E.; van Hasselt, P.M.; Haijes, H.A. Beyond nephronophthisis: Retinal dystrophy in the absence of kidney dysfunction in childhood expands the clinical spectrum of cep83 deficiency. Am. J. Med. Genet. A. 2021, 185, 2204–2210. [Google Scholar] [CrossRef]
- Alapati, A.; Goetz, K.; Suk, J.; Navani, M.; Al-Tarouti, A.; Jayasundera, T.; Tumminia, S.J.; Lee, P.; Ayyagari, R. Molecular Diagnostic Testing by eyeGENE: Analysis of Patients with Hereditary Retinal Dystrophy Phenotypes Involving Central Vision Loss. Investig. Opthalmol. Vis. Sci. 2014, 55, 5510–5521. [Google Scholar] [CrossRef] [PubMed]
- Cella, W.; Greenstein, V.C.; Zernant-Rajang, J.; Smith, T.R.; Barile, G.; Allikmets, R.; Tsang, S.H. G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull’s eye maculopathy. Exp. Eye Res. 2009, 89, 16–24. [Google Scholar] [CrossRef]
- Zhang, K.; Garibaldi, D.C.; Kniazeva, M.; Albini, T.; Chiang, M.F.; Kerrigan, M.; Sunness, J.S.; Han, M.; Allikmets, R. A novel mutation in the ABCR gene in four patients with autosomal recessive Stargardt disease. Am. J. Ophthalmol. 1999, 128, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, F.; Wang, H.; Li, Y.; Alexander, S.; Wang, K.; Willoughby, C.; Zaneveld, J.E.; Jiang, L.; Soens, Z.T.; et al. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Qual. Life Res. 2015, 134, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.J.; Nossek, C.A.; Greenberg, L.J.; Ramesar, R.S. Stargardt macular dystrophy: Common ABCA4 mutations in South Africa—establishment of a rapid genetic test and relating risk to patients. Mol. Vis. 2012, 18, 280–289. [Google Scholar] [PubMed]
- Hariri, A.H.; Gui, W.; O’Keefe, G.A.D.; Ip, M.S.; Sadda, S.R.; Gorin, M.B. Ultra-Widefield Fundus Autofluorescence Imaging of Patients with Retinitis Pigmentosa. Ophthalmol. Retin. 2018, 2, 735–745. [Google Scholar] [CrossRef]
- Wang, F.; Wang, H.; Tuan, H.-F.; Nguyen, D.H.; Sun, V.; Keser, V.; Bowne, S.J.; Sullivan, L.S.; Luo, H.; Zhao, L.; et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 2014, 133, 331–345. [Google Scholar] [CrossRef]
- Pierrache, L.H.; Hartel, B.P.; van Wijk, E.; Meester-Smoor, M.A.; Cremers, F.P.; de Baere, E.; de Zaeytijd, J.; van Schooneveld, M.J.; Cremers, C.W.; Dagnelie, G.; et al. Visual Prognosis in USH2A-Associated Retinitis Pigmentosa Is Worse for Patients with Usher Syndrome Type IIa Than for Those with Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2016, 123, 1151–1160. [Google Scholar] [CrossRef]
- Xi, Q.; Li, L.; Traboulsi, E.I.; Wang, Q.K. Novel ABCA4 compound heterozygous mutations cause severe progressive autosomal recessive cone-rod dystrophy presenting as Stargardt disease. Mol. Vis. 2009, 15, 638–645. [Google Scholar]
- Downes, S.M.; Packham, E.; Cranston, T.; Clouston, P.; Seller, A.; Németh, A.H. Detection Rate of Pathogenic Mutations in ABCA4 Using Direct Sequencing: Clinical and Research Implications. Arch. Ophthalmol. 2012, 130, 1486–1490. [Google Scholar] [CrossRef][Green Version]
- Braun, T.A.; Mullins, R.; Wagner, A.; Andorf, J.L.; Johnston, R.M.; Bakall, B.B.; DeLuca, A.; Fishman, G.A.; Lam, B.L.; Weleber, R.G.; et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet. 2013, 22, 5136–5145. [Google Scholar] [CrossRef]
- Jonsson, F.; Burstedt, M.S.; Sandgren, O.; Norberg, A.; Golovleva, I. Novel mutations in CRB1 and ABCA4 genes cause Leber congenital amaurosis and Stargardt disease in a Swedish family. Eur. J. Hum. Genet. 2013, 21, 1266–1271. [Google Scholar] [CrossRef]
- Schulz, H.L.; Grassmann, F.; Kellner, U.; Spital, G.; Rüther, K.; Jägle, H.; Hufendiek, K.; Rating, P.; Huchzermeyer, C.; Baier, M.J.; et al. Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients from a Multicenter German Cohort—Impact of Selected Deep Intronic Variants and Common SNPs. Investig. Opthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Chun, R.; Fishman, G.A.; Collison, F.T.; Stone, E.M.; Zernant, J.; Allikmets, R. The value of retinal imaging with infrared scanning laser ophthalmoscopy in patients with stargardt disease. Retina 2014, 34, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Mol. Case Stud. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Aleman, T.S.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Windsor, E.A.M.; Schwartz, S.B.; Heon, E.; Stone, E.M. Defining the Residual Vision in Leber Congenital Amaurosis Caused by RPE65 Mutations. Investig. Opthalmol. Vis. Sci. 2009, 50, 2368–2375. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Koboldt, D.C.; Bowne, S.J.; Lang, S.; Blanton, S.H.; Cadena, E.; Avery, C.E.; Lewis, R.A.; Webb-Jones, K.; Wheaton, D.H.; et al. A Dominant Mutation in Hexokinase 1 (HK1) Causes Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2014, 55, 7147–7158. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Zhang, B.; Zhao, L.; Lyubasyuk, V.; Wang, K.; Xu, M.; Li, Y.; Wu, F.; Wen, C.; et al. A Missense Mutation in HK1 Leads to Autosomal Dominant Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2014, 55, 7159–7164. [Google Scholar] [CrossRef]
- Yuan, Z.; Li, B.; Xu, M.; Chang, E.Y.; Li, H.; Yang, L.; Wu, S.; Soens, Z.T.; Li, Y.; Wong, L.-J.C.; et al. The phenotypic variability of HK1-associated retinal dystrophy. Sci. Rep. 2017, 7, 7051. [Google Scholar] [CrossRef]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Issa, P.C.; Holz, F.G.; et al. Increasing the Yield in Targeted Next-Generation Sequencing by Implicating CNV Analysis, Non-Coding Exons and the Overall Variant Load: The Example of Retinal Dystrophies. PLoS ONE 2013, 8, e78496. [Google Scholar] [CrossRef]
- Bujakowska, K.M.; Fernandez-Godino, R.; Place, E.; Consugar, M.; Navarro-Gomez, D.; White, J.; Bedoukian, E.C.; Zhu, X.; Xie, H.M.; Gai, X.; et al. Copy-number variation is an important contributor to the genetic causality of inherited retinal degenerations. Genet. Med. 2017, 19, 643–651. [Google Scholar] [CrossRef]
- Abu-Safieh, L.; Vithana, E.N.; Mantel, I.; Holder, G.E.; Pelosini, L.; Bird, A.C.; Bhattacharya, S.S. A large deletion in the adRP gene PRPF31: Evidence that haploinsufficiency is the cause of disease. Mol. Vis. 2006, 12, 384–388. [Google Scholar]
- Köhn, L.M.; Bowne, S.J.; Sullivan, L.S.; Daiger, S.P.; Burstedt, M.S.I.; Kadzhaev, K.; Sandgren, O.; Golovleva, I. Breakpoint characterization of a novel ∼59 kb genomic deletion on 19q13.42 in autosomal-dominant retinitis pigmentosa with incomplete penetrance. Eur. J. Hum. Genet. 2009, 17, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; De Pozo-Valero, M.L.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Anesth. Analg. 2020, 22, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Ecob, R.; Costello, H.; Sweeney, M.G.; Duncan, A.J.; Pearce, K.; Strachan, D.; Forge, A.; Davis, A.; Bitner-Glindzicz, M. Hearing in 44–45 year olds with m.1555A>G, a genetic mutation predisposing to aminoglycoside-induced deafness: A population based cohort study. BMJ Open 2012, 2, e000411. [Google Scholar] [CrossRef]
- Jing, W.; Zongjie, H.; Denggang, F.; Na, H.; Bin, Z.; Aifen, Z.; Xijiang, H.; Cong, Y.; Yunping, D.; Ring, H.Z.; et al. Mitochondrial mutations associated with aminoglycoside ototoxicity and hearing loss susceptibility identified by meta-analysis. J. Med. Genet. 2015, 52, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Estivill, X.; Govea, N.; Barceló, A.; Perelló, E.; Badenas, C.; Romero, E.; Moral, L.; Scozzari, R.; D’Urbano, L.; Zeviani, M.; et al. Familial Progressive Sensorineural Deafness Is Mainly Due to the mtDNA A1555G Mutation and Is Enhanced by Treatment with Aminoglycosides. Am. J. Hum. Genet. 1998, 62, 27–35. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Barbaro, V.; Migliorati, A.; Raffa, P.; Nespeca, P.; De Nadai, K.; DEL Vecchio, C.; Palù, G.; Parolin, C.; Di Iorio, E. Novel Variants of RPGR in X-Linked Retinitis Pigmentosa Families and Genotype-Phenotype Correlation. Eur. J. Ophthalmol. 2017, 27, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Miano, M.G.; Testa, F.; Filippini, F.; Trujillo, M.; Conte, I.; Lanzara, C.; Millán, J.M.; De Bernardo, C.; Grammatico, B.; Mangino, M.; et al. Identification of novel RP2 mutations in a subset of X-linked retinitis pigmentosa families and prediction of new domains. Hum. Mutat. 2001, 18, 109–119. [Google Scholar] [CrossRef]
- Kurata, K.; Hosono, K.; Hayashi, T.; Mizobuchi, K.; Katagiri, S.; Miyamichi, D.; Nishina, S.; Sato, M.; Azuma, N.; Nakano, T.; et al. X-linked Retinitis Pigmentosa in Japan: Clinical and Genetic Findings in Male Patients and Female Carriers. Int. J. Mol. Sci. 2019, 20, 1518. [Google Scholar] [CrossRef]
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef]
- Audo, I.; Bujakowska, K.M.; Léveillard, T.; Mohand-Saïd, S.; Lancelot, M.-E.; Germain, A.; Antonio, A.; Michiels, C.; Saraiva, J.-P.; Letexier, M.; et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet. J. Rare Dis. 2012, 7, 8. [Google Scholar] [CrossRef]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.; Mullaney, B.G.; Bhaskar, S.S.; Dickerson, J.E.; Hall, G.; O’Grady, A.; Webster, A.; Ramsden, S.C.; Black, G.C. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J. Med. Genet. 2012, 49, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Corton, M.; Nishiguchi, K.M.; Avila-Fernandez, A.; Nikopoulos, K.; Riveiro-Álvarez, R.; Tatu, S.D.; Ayuso, C.; Rivolta, C. Exome Sequencing of Index Patients with Retinal Dystrophies as a Tool for Molecular Diagnosis. PLoS ONE 2013, 8, e65574. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.-H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef]
- Sheck, L.H.N.; Esposti, S.D.; Mahroo, O.A.; Arno, G.; Pontikos, N.; Wright, G.; Webster, A.R.; Khan, K.N.; Michaelides, M. Panel-based genetic testing for inherited retinal disease screening 176 genes. Mol. Genet. Genom. Med. 2021, 9, e1663. [Google Scholar] [CrossRef]
- Cremers, F.P.; Lee, W.; Collin, R.W.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef]
- Del Pozo-Valero, M.; Riveiro-Alvarez, R.; Martin-Merida, I.; Blanco-Kelly, F.; Swafiri, S.; Lorda-Sanchez, I.; Trujillo-Tiebas, M.J.; Carreño, E.; Jimenez-Rolando, B.; Garcia-Sandoval, B.; et al. Impact of Next Generation Sequencing in Unraveling the Genetics of 1036 Spanish Families with Inherited Macular Dystrophies. Investig. Ophthalmol. Vis. Sci. 2022, 63, 11. [Google Scholar] [CrossRef]
- Roosing, S.; Thiadens, A.A.; Zekveld-Vroon, R.C.; Hoyng, C.B.; Hollander, A.I.d.; Cremers, F.P.; Klaver, C.C. Is there evidence for a digenic model in stargardt disease? Investig. Ophthalmol. Vis. Sci. 2011, 52, 5395. [Google Scholar]
- Zhou, R.; Mawatari, G.; Cai, X.-B.; Shen, R.-J.; Wang, Y.-H.; Guo, Y.-M.; Guo, F.-Y.; Yuan, J.; Pan, D.; Nao-I, N.; et al. CLEC3B is a novel causative gene for macular-retinal dystrophy. Genet. Med. 2022, 24, 1249–1260. [Google Scholar] [CrossRef]
- Bartolini, F.; Bhamidipati, A.; Thomas, S.; Schwahn, U.; Lewis, S.A.; Cowan, N.J. Functional Overlap between Retinitis Pigmentosa 2 Protein and the Tubulin-specific Chaperone Cofactor C. J. Biol. Chem. 2002, 277, 14629–14634. [Google Scholar] [CrossRef]
- Kühnel, K.; Veltel, S.; Schlichting, I.; Wittinghofer, A. Crystal Structure of the Human Retinitis Pigmentosa 2 Protein and Its Interaction with Arl3. Structure 2006, 14, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9, a028191. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef]
- Thomas, S.; Boutaud, L.; Reilly, M.L.; Benmerah, A. Cilia in hereditary cerebral anomalies. Biol. Cell 2019, 111, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesth. Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).