
Exome-Wide Association Study Identified Clusters of Pleiotropic Genetic Associations with Alzheimer’s Disease and Thirteen Cardiovascular Traits


Abstract
:1. Introduction
2. Materials and Methods
2.1. Accession Numbers
2.2. Study Cohorts
2.3. Genotypes
2.4. Phenotypes
2.5. Correlations among Phenotypes
2.6. Correlations among Summary Statistics
2.7. Statistical Analyses
2.8. Pleiotropic Associations
2.9. Index SNPs and Gene Mapping
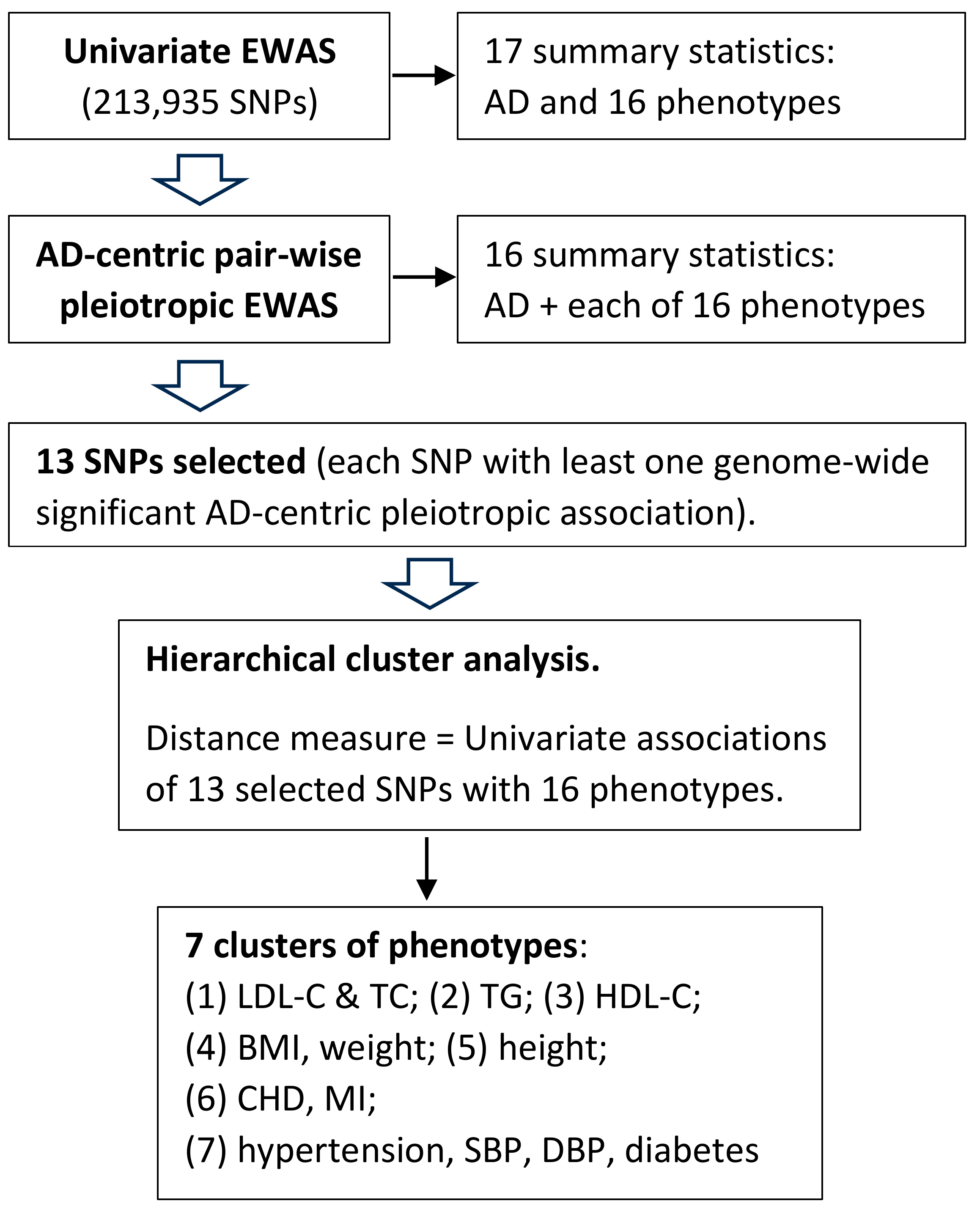
2.10. Cluster Analysis
3. Results
3.1. Univariate Associations from EWAS
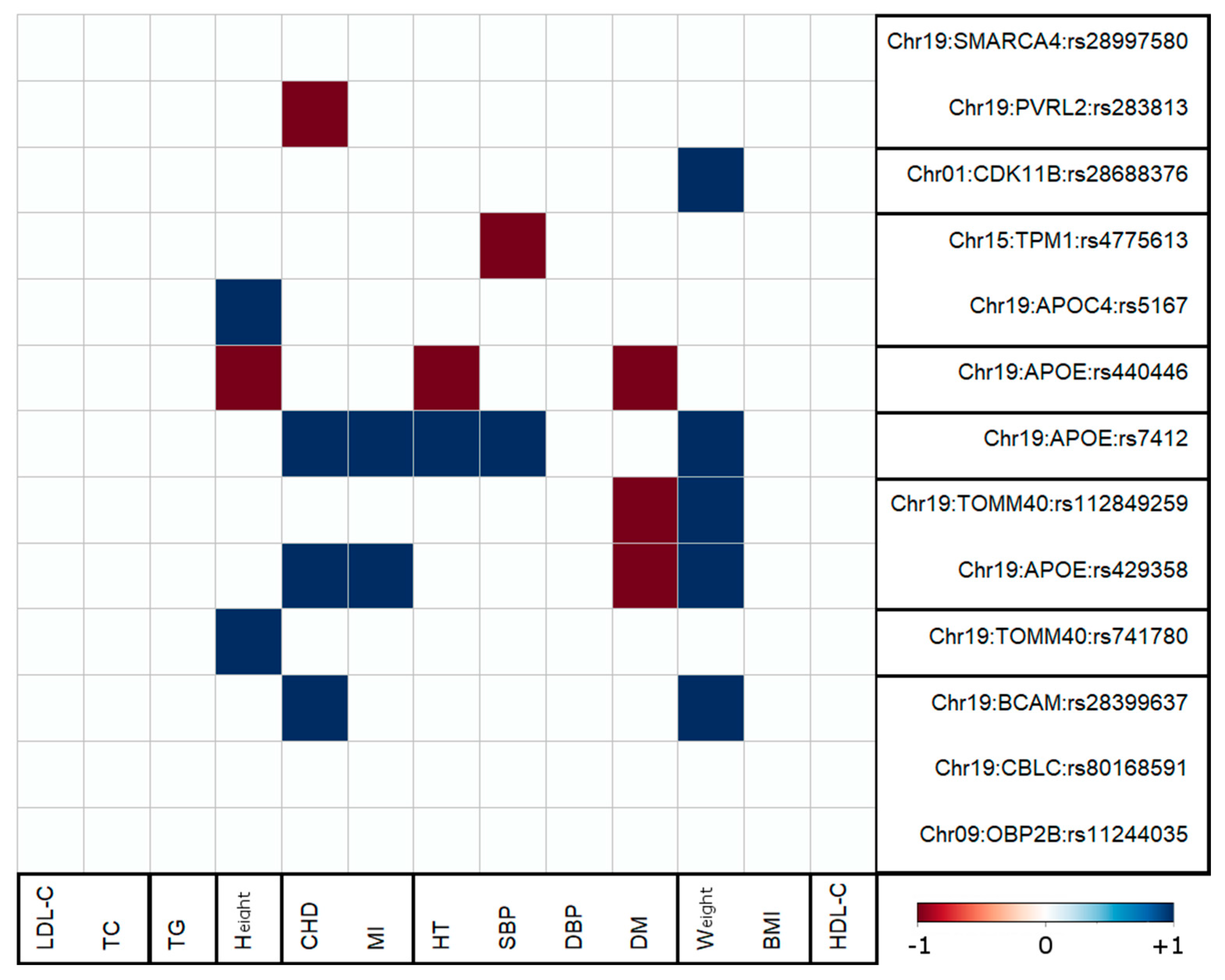
3.2. Pleiotropic AD-Centric Pair-Wise Associations
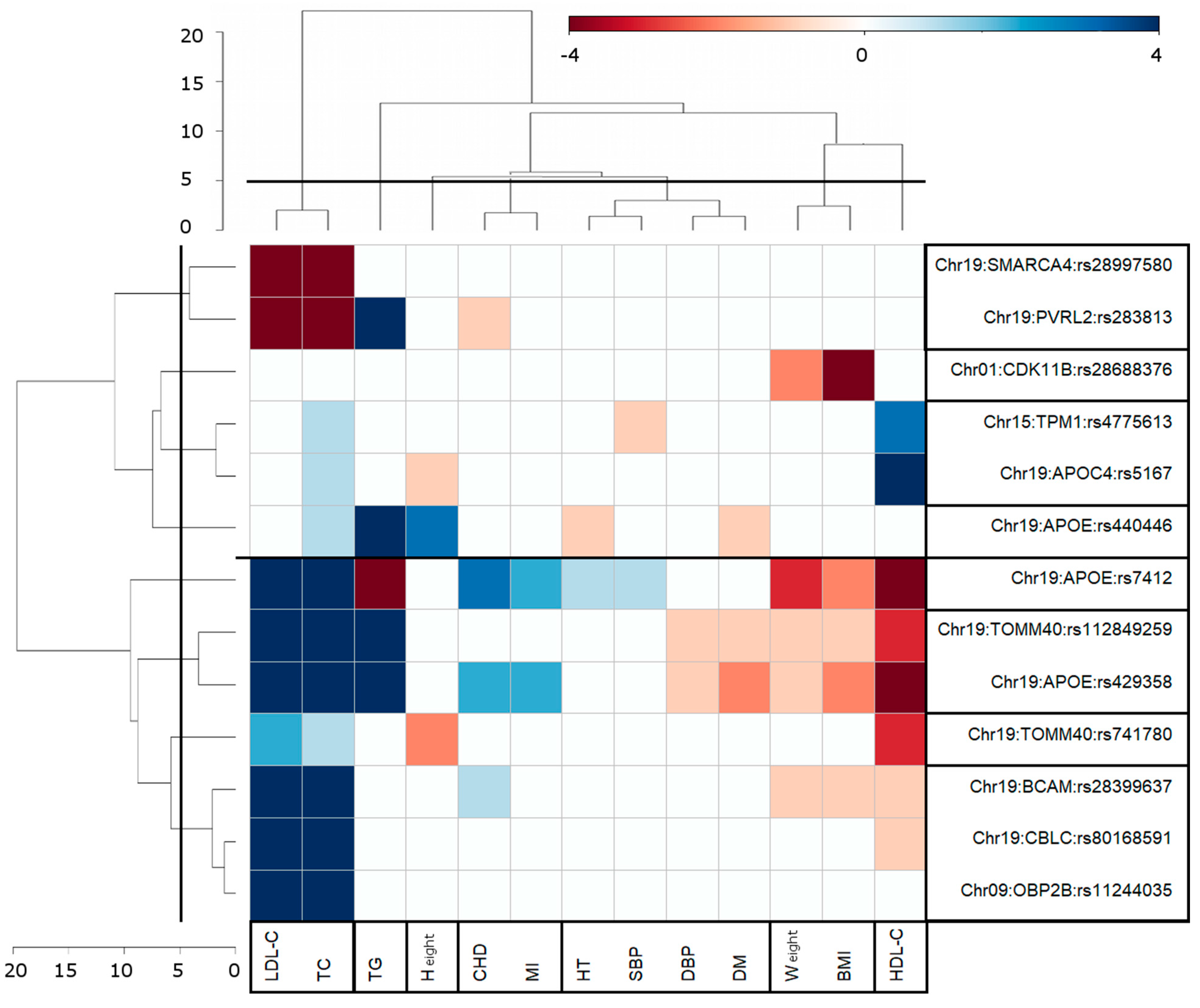
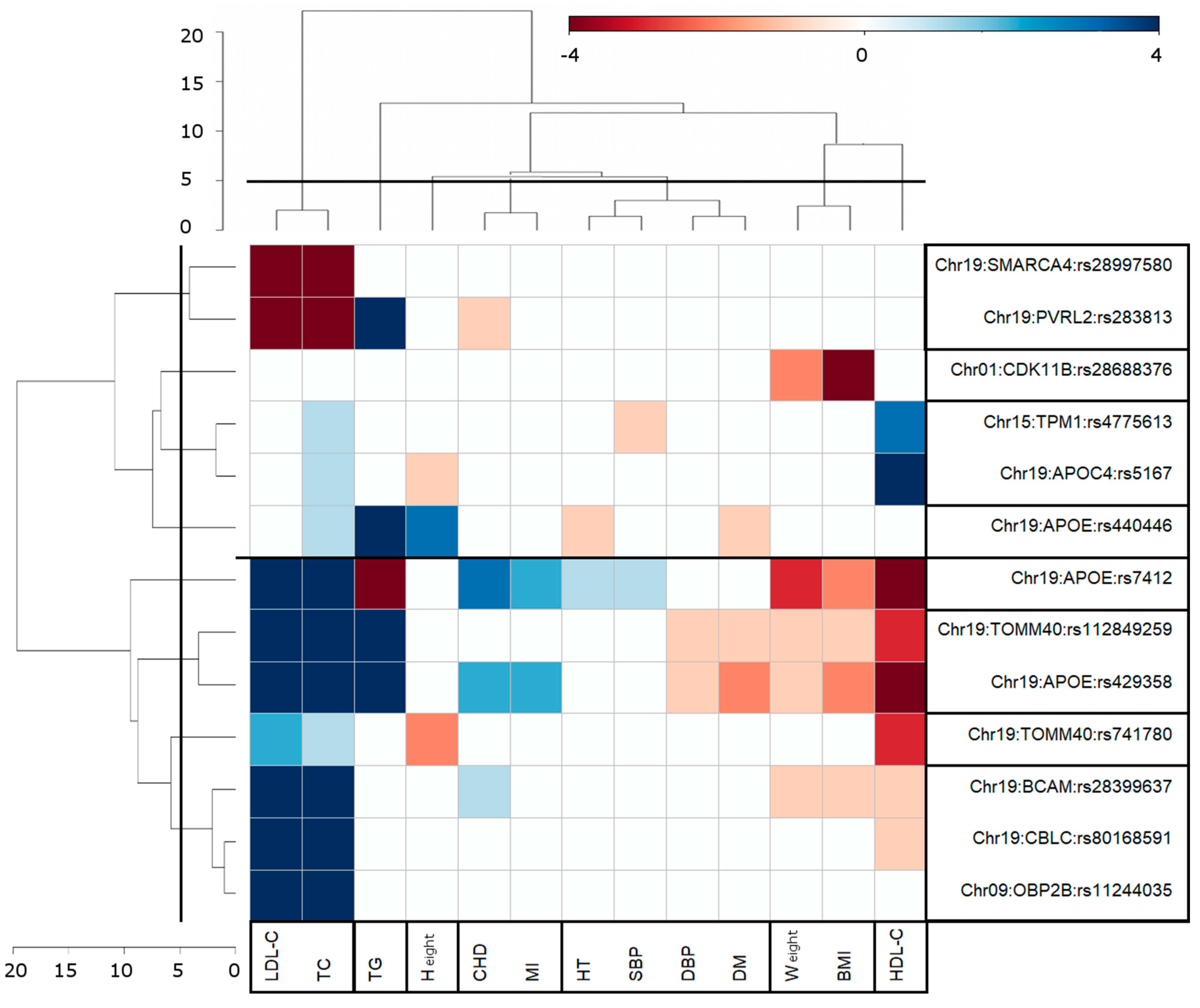
3.3. Clusters of Pleiotropic Associations
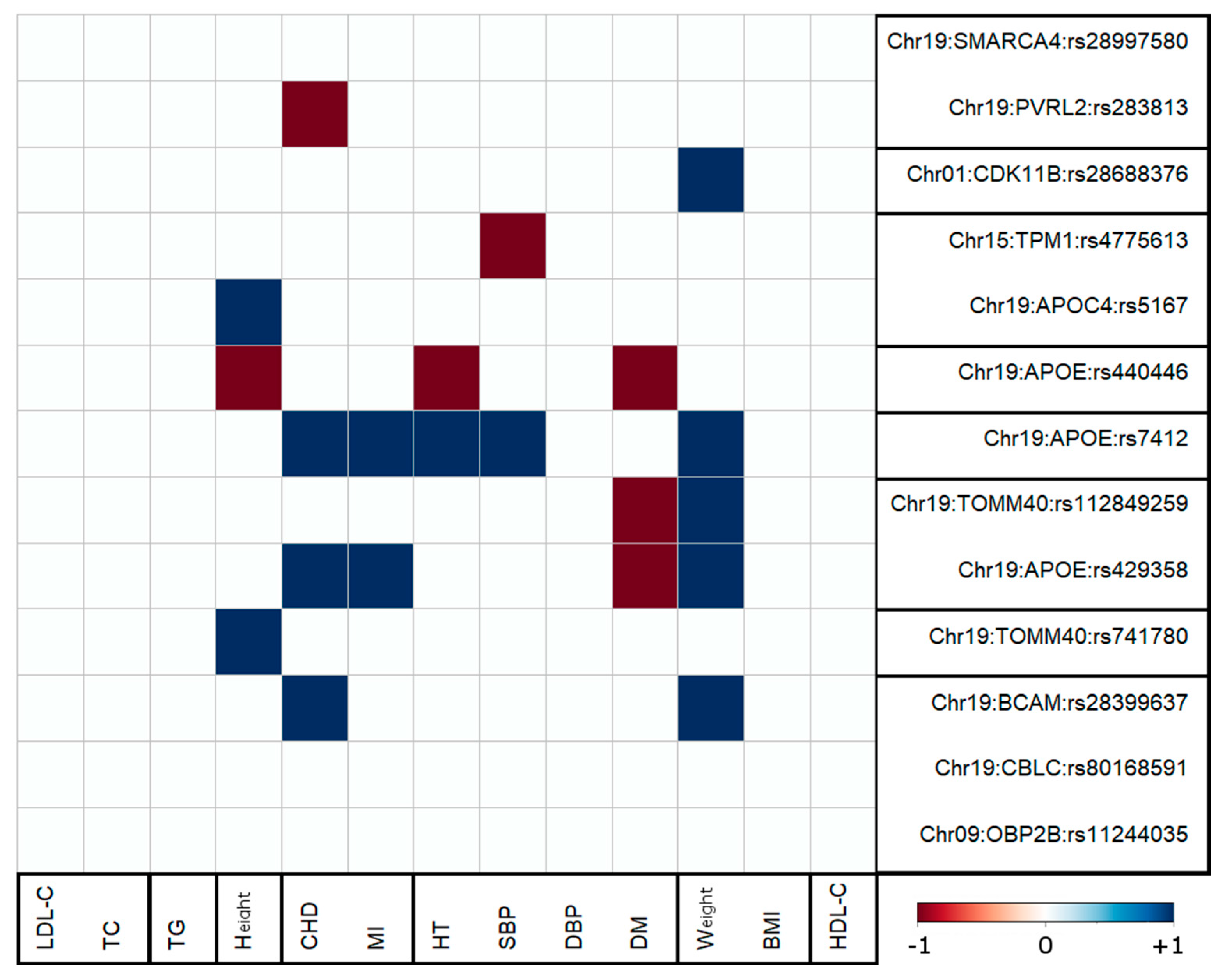
3.4. Antagonistic Genetic Heterogeneity
4. Discussion
4.1. AD-Centric Pair-Wise Pleiotropic Associations
4.2. Clustering of Genome-Wide Significant Pair-Wise AD-Centric Pleiotropic Associations
4.3. Antagonistic Genetic Heterogeneity Was Observed for AD-Centric Pleiotropic Associations with Five Traits
4.4. Protective Effect of Higher BMI Level against AD Is Related to High Weight
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gaugler, J.; James, B.; Johnson, T.; Reimer, J.; Solis, M.; Weuve, J. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Malik, R.; Kalra, S.; Bhatia, S.; Harrasi, A.A.; Singh, G.; Mohan, S.; Makeen, H.A.; Albratty, M.; Meraya, A.; Bahar, B.; et al. Overview of therapeutic targets in management of dementia. Biomed. Pharmacother. 2022, 152, 113168. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J. Anti-Amyloid Monoclonal Antibodies are Transformative Treatments that Redefine Alzheimer’s Disease Therapeutics. Drugs 2023, 83, 569–576. [Google Scholar] [CrossRef]
- Ferrer, I. Hypothesis review: Alzheimer’s overture guidelines. Brain Pathol. 2023, 33, e13122. [Google Scholar] [CrossRef]
- Pons, V.; Rivest, S. Targeting Systemic Innate Immune Cells as a Therapeutic Avenue for Alzheimer Disease. Pharmacol. Rev. 2022, 74, 1–17. [Google Scholar] [CrossRef]
- Tang, Q.; Wang, F.; Yang, J.; Peng, H.; Li, Y.; Li, B.; Wang, S. Revealing a Novel Landscape of the Association Between Blood Lipid Levels and Alzheimer’s Disease: A Meta-Analysis of a Case-Control Study. Front. Aging Neurosci. 2019, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Anstey, K.J.; Ashby-Mitchell, K.; Peters, R. Updating the Evidence on the Association between Serum Cholesterol and Risk of Late-Life Dementia: Review and Meta-Analysis. J. Alzheimer’s Dis. 2017, 56, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tong, T.; Chang, A.; Ang, T.F.A.; Tao, Q.; Auerbach, S.; Devine, S.; Qiu, W.Q.; Mez, J.; Massaro, J.; et al. Midlife lipid and glucose levels are associated with Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 181–193. [Google Scholar] [CrossRef]
- Bone, W.P.; Siewert, K.M.; Jha, A.; Klarin, D.; Damrauer, S.M.; Program, V.A.M.V.; Chang, K.M.; Tsao, P.S.; Assimes, T.L.; Ritchie, M.D.; et al. Multi-trait association studies discover pleiotropic loci between Alzheimer’s disease and cardiometabolic traits. Alzheimer’s Res. Ther. 2021, 13, 34. [Google Scholar] [CrossRef]
- Broce, I.J.; Tan, C.H.; Fan, C.C.; Jansen, I.; Savage, J.E.; Witoelar, A.; Wen, N.; Hess, C.P.; Dillon, W.P.; Glastonbury, C.M.; et al. Dissecting the genetic relationship between cardiovascular risk factors and Alzheimer’s disease. Acta Neuropathol. 2019, 137, 209–226. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Reitz, C.; Honig, L.S.; Tang, M.X.; Shea, S.; Mayeux, R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005, 65, 545–551. [Google Scholar] [CrossRef]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef]
- Xiang, Q.; Andersen, S.L.; Sweigart, B.; Gunn, S.; Nygaard, M.; Perls, T.T.; Sebastiani, P. Signatures of Neuropsychological Test Results in the Long Life Family Study: A Cluster Analysis. J. Alzheimer’s Dis. 2023, 93, 1457–1469. [Google Scholar] [CrossRef]
- Ramoni, M.F.; Sebastiani, P.; Kohane, I.S. Cluster analysis of gene expression dynamics. Proc. Natl. Acad. Sci. USA 2002, 99, 9121–9126. [Google Scholar] [CrossRef] [PubMed]
- Bondi, M.W.; Edmonds, E.C.; Jak, A.J.; Clark, L.R.; Delano-Wood, L.; McDonald, C.R.; Nation, D.A.; Libon, D.J.; Au, R.; Galasko, D.; et al. Neuropsychological criteria for mild cognitive impairment improves diagnostic precision, biomarker associations, and progression rates. J. Alzheimer’s Dis. 2014, 42, 275–289. [Google Scholar] [CrossRef]
- Edmonds, E.C.; McDonald, C.R.; Marshall, A.; Thomas, K.R.; Eppig, J.; Weigand, A.J.; Delano-Wood, L.; Galasko, D.R.; Salmon, D.P.; Bondi, M.W.; et al. Early versus late MCI: Improved MCI staging using a neuropsychological approach. Alzheimer’s Dement. 2019, 15, 699–708. [Google Scholar] [CrossRef]
- Jak, A.J.; Preis, S.R.; Beiser, A.S.; Seshadri, S.; Wolf, P.A.; Bondi, M.W.; Au, R. Neuropsychological Criteria for Mild Cognitive Impairment and Dementia Risk in the Framingham Heart Study. J. Int. Neuropsychol. Soc. 2016, 22, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Kulminski, A.M. Genetic Association Studies of Age-Related Traits: New Perspectives. Adv. Geriatr. Med. Res. 2021, 3, e210003. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.A.; Urbanowicz, R.J.; Naj, A.C.; Moore, J.H. Genetic heterogeneity: Challenges, impacts, and methods through an associative lens. Genet. Epidemiol. 2022, 46, 555–571. [Google Scholar] [CrossRef]
- Pedditzi, E.; Peters, R.; Beckett, N. The risk of overweight/obesity in mid-life and late life for the development of dementia: A systematic review and meta-analysis of longitudinal studies. Age Ageing 2016, 45, 14–21. [Google Scholar] [CrossRef]
- Tolppanen, A.M.; Ngandu, T.; Kareholt, I.; Laatikainen, T.; Rusanen, M.; Soininen, H.; Kivipelto, M. Midlife and late-life body mass index and late-life dementia: Results from a prospective population-based cohort. J. Alzheimer’s Dis. 2014, 38, 201–209. [Google Scholar] [CrossRef]
- Kulminski, A.M.; Loika, Y.; Huang, J.; Arbeev, K.G.; Bagley, O.; Ukraintseva, S.; Yashin, A.I.; Culminskaya, I. Pleiotropic Meta-Analysis of Age-Related Phenotypes Addressing Evolutionary Uncertainty in Their Molecular Mechanisms. Front. Genet. 2019, 10, 433. [Google Scholar] [CrossRef]
- Kulminski, A.M.; Huang, J.; Loika, Y.; Arbeev, K.G.; Bagley, O.; Yashkin, A.; Duan, M.; Culminskaya, I. Strong impact of natural-selection-free heterogeneity in genetics of age-related phenotypes. Aging 2018, 10, 492–514. [Google Scholar] [CrossRef]
- Kulminski, A.M.; Loika, Y.; Nazarian, A.; Culminskaya, I. Quantitative and Qualitative Role of Antagonistic Heterogeneity in Genetics of Blood Lipids. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2020, 75, 1811–1819. [Google Scholar] [CrossRef]
- Loika, Y.; Feng, F.; Loiko, E.; Kulminski, A.M. Mediation of the APOE associations with Alzheimer’s and coronary heart diseases through body mass index and lipids. GeroScience 2022, 44, 1141–1156. [Google Scholar] [CrossRef] [PubMed]
- Kulminski, A.M.; Loiko, E.; Loika, Y.; Culminskaya, I. Pleiotropic predisposition to Alzheimer’s disease and educational attainment: Insights from the summary statistics analysis. GeroScience 2022, 44, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Kulminski, A.M.; Feng, F.; Loiko, E.; Nazarian, A.; Loika, Y.; Culminskaya, I. Prevailing Antagonistic Risks in Pleiotropic Associations with Alzheimer’s Disease and Diabetes. J. Alzheimer’s Dis. 2023, 94, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Szustakowski, J.D.; Balasubramanian, S.; Kvikstad, E.; Khalid, S.; Bronson, P.G.; Sasson, A.; Wong, E.; Liu, D.; Wade Davis, J.; Haefliger, C.; et al. Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat. Genet. 2021, 53, 942–948. [Google Scholar] [CrossRef]
- Fisher, R.A.S. Statistical Methods for Research Workers, 14th ed.; Oliver and Boyd: Edinburgh, UK, 1970. [Google Scholar]
- Leslie, R.; O’Donnell, C.J.; Johnson, A.D. GRASP: Analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics 2014, 30, i185–i194. [Google Scholar] [CrossRef]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Gunes, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and deposition resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef] [PubMed]
- Bajic, V.P.; Su, B.; Lee, H.G.; Kudo, W.; Siedlak, S.L.; Zivkovic, L.; Spremo-Potparevic, B.; Djelic, N.; Milicevic, Z.; Singh, A.K.; et al. Mislocalization of CDK11/PITSLRE, a regulator of the G2/M phase of the cell cycle, in Alzheimer disease. Cell. Mol. Biol. Lett. 2011, 16, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Fredi, B.M.; De Labio, R.W.; Rasmussen, L.T.; Chagas, E.F.B.; Chen, E.S.; Turecki, G.; Smith, M.A.C.; Payao, S.L.M. CDK10, CDK11, FOXO1, and FOXO3 Gene Expression in Alzheimer’s Disease Encephalic Samples. Cell. Mol. Neurobiol. 2023, 43, 2953–2962. [Google Scholar] [CrossRef]
- Choi, H.H.; Choi, H.K.; Jung, S.Y.; Hyle, J.; Kim, B.J.; Yoon, K.; Cho, E.J.; Youn, H.D.; Lahti, J.M.; Qin, J.; et al. CHK2 kinase promotes pre-mRNA splicing via phosphorylating CDK11(p110). Oncogene 2014, 33, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.; Sekiya, M.; Taniguchi, T.; Iijima, K.M.; Wang, R.; Ando, K. Global analysis of phosphorylation of tau by the checkpoint kinases Chk1 and Chk2 in vitro. J. Proteome Res. 2013, 12, 2654–2665. [Google Scholar] [CrossRef]
- Guven, K.; Gunning, P.; Fath, T. TPM3 and TPM4 gene products segregate to the postsynaptic region of central nervous system synapses. Bioarchitecture 2011, 1, 284–289. [Google Scholar] [CrossRef]
- Pelucchi, S.; Stringhi, R.; Marcello, E. Dendritic Spines in Alzheimer’s Disease: How the Actin Cytoskeleton Contributes to Synaptic Failure. Int. J. Mol. Sci. 2020, 21, 908. [Google Scholar] [CrossRef]
- Galloway, P.G.; Mulvihill, P.; Siedlak, S.; Mijares, M.; Kawai, M.; Padget, H.; Kim, R.; Perry, G. Immunochemical demonstration of tropomyosin in the neurofibrillary pathology of Alzheimer’s disease. Am. J. Pathol. 1990, 137, 291–300. [Google Scholar]
- Reumiller, C.M.; Schmidt, G.J.; Dhrami, I.; Umlauf, E.; Rappold, E.; Zellner, M. Gender-related increase of tropomyosin-1 abundance in platelets of Alzheimer’s disease and mild cognitive impairment patients. J. Proteom. 2018, 178, 73–81. [Google Scholar] [CrossRef]
- Castano, E.M.; Maarouf, C.L.; Wu, T.; Leal, M.C.; Whiteside, C.M.; Lue, L.F.; Kokjohn, T.A.; Sabbagh, M.N.; Beach, T.G.; Roher, A.E. Alzheimer disease periventricular white matter lesions exhibit specific proteomic profile alterations. Neurochem. Int. 2013, 62, 145–156. [Google Scholar] [CrossRef] [PubMed]
- England, J.; Granados-Riveron, J.; Polo-Parada, L.; Kuriakose, D.; Moore, C.; Brook, J.D.; Rutland, C.S.; Setchfield, K.; Gell, C.; Ghosh, T.K.; et al. Tropomyosin 1: Multiple roles in the developing heart and in the formation of congenital heart defects. J. Mol. Cell. Cardiol. 2017, 106, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Receptor-mediated endocytosis: Insights from the lipoprotein receptor system. Proc. Natl. Acad. Sci. USA 1979, 76, 3330–3337. [Google Scholar] [CrossRef]
- Gouveia, C.; Gibbons, E.; Dehghani, N.; Eapen, J.; Guerreiro, R.; Bras, J. Genome-wide association of polygenic risk extremes for Alzheimer’s disease in the UK Biobank. Sci. Rep. 2022, 12, 8404. [Google Scholar] [CrossRef]
- Gallo, S.; Byham-Gray, L.; Duffy, V.B.; Hoffman, H.J.; Hayes, J.E.; Rawal, S. Associations of olfactory dysfunction with anthropometric and cardiometabolic measures: Findings from the 2013–2014 national health and nutrition examination survey (NHANES). Physiol. Behav. 2020, 215, 112702. [Google Scholar] [CrossRef] [PubMed]
- Loika, Y.; Loiko, E.; Feng, F.; Stallard, E.; Yashin, A.I.; Arbeev, K.; Kuipers, A.L.; Feitosa, M.F.; Province, M.A.; Kulminski, A.M. Exogenous exposures shape genetic predisposition to lipids, Alzheimer’s, and coronary heart disease in the MLXIPL gene locus. Aging 2023, 15, 3249–3272. [Google Scholar] [CrossRef]
- Hao, K.; Di Narzo, A.F.; Ho, L.; Luo, W.; Li, S.; Chen, R.; Li, T.; Dubner, L.; Pasinetti, G.M. Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Mol. Asp. Med. 2015, 43–44, 66–76. [Google Scholar] [CrossRef]
- Hardy, J.; de Strooper, B.; Escott-Price, V. Diabetes and Alzheimer’s disease: Shared genetic susceptibility? Lancet Neurol. 2022, 21, 962–964. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| AD Status | PAR | N | Women | BC Range | LS | Age | Mortality | |||
|---|---|---|---|---|---|---|---|---|---|---|
| VALUE (SD) | 188,260 | 103,736 (55.10) | 1936–1970 | 65.21 (8.0) | 56.70 (8.03) | 899 (0.48) | ||||
| Quantitative traits | ||||||||||
| AD | DM | HT | CHD | MI | STROKE | HF | ||||
| Cases | 361 | 53 (15%) | 189 (52%) | 89 (25%) | 37 (10%) | 30 (8%) | 20 (6%) | |||
| Controls | 187,899 | 11,015 (6%) | 45,349 (24%) | 16,327 (9%) | 5834 (3%) | 4589 (2%) | 3236 (2%) | |||
| p-value | 1.33 × 10−9 | <2.2 × 10−16 | <2.2 × 10−16 | 3.90 × 10−10 | 9.97 × 10−9 | 6.867 × 10−6 | ||||
| Quantitative traits | ||||||||||
| BG (mg/dL) | BMI (kg/m2) | Height (cm) | Weight (kg) | SBP (mmHg) | DBP (mmHg) | HDL-C (mg/dL) | LDL-C (mg/dL) | TC (mg/dL) | TG (mg/dL) | |
| Cases | 93.36 (19.93) | 27.01 (4.90) | 167.18 (9.17) | 75.62 (15.40) | 146.18 (19.58) | 81.79 (10.96) | 57.77 (16.30) | 133.77 (35.72) | 216.97 (47.43) | 149.26 (78.77) |
| Controls | 92.13 (21.19) | 27.34 (4.73) | 168.67 (9.24) | 78.03 (15.83) | 139.78 (19.58) | 82.13 (10.65) | 56.49 (14.82) | 138.07 (33.44) | 221.19 (43.99) | 154.26 (89.82) |
| p-value | 5.89 × 10−1 | 5.05 × 10−1 | 1.61 × 10−1 | 1.72 × 10−1 | 3.46 × 10−2 | 9.04 × 10−1 | 4.77 × 10−1 | 2.68 × 10−1 | 3.96 × 10−1 | 5.54 × 10−1 |
| Individual-Level Data | Summary Statistics | |||
|---|---|---|---|---|
| Phenotype | r | P | r | P |
| HT * | 0.0290 | 2.41 × 10−23 | 0.0352 | <2.2 × 10−16 |
| CHD * | 0.0248 | 1.82 × 10−17 | 0.0145 | 2.15 × 10−11 |
| MI * | 0.0180 | 6.52 × 10−10 | −0.0169 | 4.67 × 10−15 |
| STROKE * | 0.0166 | 1.19 × 10−8 | −0.0174 | 8.10 × 10−16 |
| DM * | 0.0164 | 1.76 × 10−8 | 0.0365 | <2.2 × 10−16 |
| HF * | 0.0128 | 1.08 × 10−5 | −0.0358 | <2.2 × 10−16 |
| BG | 0.0025 | 3.82 × 10−1 | −0.0069 | 1.55 × 10−3 |
| BMI | −0.0031 | 2.82 × 10−1 | 0.0514 | <2.2 × 10−16 |
| Height * | −0.0070 | 1.61 × 10−2 | −0.0700 | <2.2 × 10−16 |
| Weight * | −0.0066 | 2.30 × 10−2 | 0.0092 | 2.03 × 10−5 |
| SBP * | 0.0143 | 9.34 × 10−7 | −0.0082 | 1.41 × 10−4 |
| DBP | −0.0014 | 6.35 × 10−1 | 0.0164 | 2.87 × 10−14 |
| HDL-C | 0.0038 | 1.96 × 10−1 | −0.0074 | 5.84 × 10−4 |
| LDL-C | −0.0057 | 5.17 × 10−2 | −0.0029 | 1.87 × 10−1 |
| TC | −0.0042 | 1.48 × 10−1 | −0.0086 | 7.62 × 10−5 |
| TG | −0.0024 | 4.00 × 10−1 | −0.0078 | 3.41 × 10−4 |
| N | Gene(s) 1 | SNP 2 | Chr | Location, Base Pairs GRCh38 | Ref /Alt | MAF obs (%) | PHWE | Function | Beta | SE | P | Gene GRASP | SNP GRASP | r2 | PGRASP | PMID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CDK11B (MMP23B) | rs28688376 | 1 | 1,637,577 | T/c | 26.1 | 6 × 10−36 | intron | 0.298 | 0.079 | 1.56 × 10−4 | |||||
| 2 | OBP2B | rs11244035 | 9 | 133,205,932 | C/t | 10.2 | 2 × 10−30 | missense | 0.393 | 0.109 | 3.23 × 10−4 | ABO | rs8176694 | 0.02 | 1.3 × 10−2 | 20061627 |
| 3 | TPM1 | rs4775613 | 15 | 63,056,897 | A/g | 42.8 | 3 × 10−2 | 5′UTR | −0.334 | 0.078 | 1.99 × 10−5 | RAB8B | rs10519190 | 0.00 | 2.2 × 10−4 | 17998437 |
| 4 | SMARCA4 | rs28997580 | 19 | 11,013,062 | C/t | 0.9 | 3 × 10−2 | synonymous | 0.960 | 0.255 | 1.64 × 10−4 | LDLR | rs2569540 | 0.00 | 1 × 10−9 | 35589863 |
| APOE | 19 | |||||||||||||||
| 5 | CBLC | rs80168591 | 19 | 44,781,370 | G/a | 1.4 | 0.87 | splice | 0.911 | 0.205 | 8.54 × 10−6 | CBLC | rs899087 | 0.00 | 5.1 × 10−5 | 22832961 |
| 6 | BCAM | rs28399637 | 19 | 44,820,881 | G/a | 31.5 | 5 × 10−8 | intron | 0.382 | 0.076 | 4.71 × 10−7 | BCAM | rs2927480 | 0.21 | 5.0 × 10−49 | 21460841 |
| 7 | PVRL2 | rs283813 | 19 | 44,885,917 | T/a | 6.7 | 3 × 10−22 | intron | 0.488 | 0.123 | 7.16 × 10−5 | PVRL2 | rs283813 | 1 | 7.6 × 10−28 | 33589840 |
| 8 | TOMM40 | rs112849259 | 19 | 44,894,050 | C/t | 2.6 | 0.73 | missense | 1.227 | 0.138 | 4.59 × 10−19 | |||||
| 9 | TOMM40 | rs741780 | 19 | 44,901,174 | T/c | 43.2 | 7 × 10−2 | intron | −0.454 | 0.080 | 1.14 × 10−8 | TOMM40 | rs741780 | 1 | 1.4 × 10−8 | 23565137 |
| 10 | APOE | rs440446 | 19 | 44,905,910 | G/c | 35.8 | 2 × 10−2 | missense | −0.561 | 0.087 | 1.22 × 10−10 | APOE | rs439401 | 0.57 | 1.1 × 10−78 | 21460841 |
| 11 | APOE | rs429358 | 19 | 44,908,684 | T/c | 15.4 | 0.54 | missense | 1.371 | 0.077 | 4.63 × 10−71 | APOE | rs429358 | 1 | 2.7 × 10−78 | 21390209 |
| 12 | APOE | rs7412 | 19 | 44,908,822 | C/t | 8.0 | 0.66 | missense | −0.856 | 0.200 | 1.87 × 10−5 | APOE | rs7412 | 1 | 5.5 × 10−58 | 20885792 |
| 13 | APOC4; APOC2 | rs5167 | 19 | 44,945,208 | T/g | 35.1 | 0.10 | missense | 0.315 | 0.075 | 2.92 × 10−5 | APOC4; APOC2 | rs5167 | 1 | 2.8 × 10−9 | 21460840 |
| N | Gene(s) 1 | SNP 2 | Chr | Location, Base Pairs GRCh38 | Ref /Alt | MAF obs (%) | PH | Beta | SE | P | PF |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CDK11B (MMP23B) | rs28688376 | 1 | 1,637,577 | T/c | 26.1 | BMI | −0.096 | 0.017 | 3.36 × 10−8 | 1.42 × 10−10 |
| 2 | CDK11B (MMP23B) | rs28688376 | 1 | 1,637,577 | T/c | 26.1 | Weight | −0.232 | 0.052 | 7.01 × 10−6 | 2.37 × 10−8 |
| 3 | OBP2B | rs11244035 | 9 | 133,205,932 | C/t | 10.2 | LDL-C | 1.206 | 0.187 | 1.09 × 10−10 | 1.13 × 10−12 |
| 4 | OBP2B | rs11244035 | 9 | 133,205,932 | C/t | 10.2 | TC | 1.519 | 0.243 | 3.84 × 10−10 | 3.81 × 10−12 |
| 5 | TPM1 | rs4775613 | 15 | 63,056,897 | A/g | 42.8 | TC | −0.436 | 0.146 | 2.81 × 10−3 | 9.91 × 10−7 |
| 6 | TPM1 | rs4775613 | 15 | 63,056,897 | A/g | 42.8 | HDL-C | −0.245 | 0.048 | 2.43 × 10−7 | 1.31 × 10−10 |
| 7 | TPM1 | rs4775613 | 15 | 63,056,897 | A/g | 42.8 | SBP | 0.156 | 0.062 | 1.18 × 10−2 | 3.84 × 10−6 |
| 8 | TPM1 | rs4775613 | 15 | 63,056,897 | A/g | 42.8 | HF | −0.052 | 0.026 | 4.16 × 10−2 | 1.24 × 10−5 |
| 9 | SMARCA4 | rs28997580 | 19 | 11,013,062 | C/t | 0.9 | LDL-C | −5.450 | 0.591 | 2.81 × 10−20 | <5 × 10−8 |
| 10 | SMARCA4 | rs28997580 | 19 | 11,013,062 | C/t | 0.9 | TC | −6.141 | 0.767 | 1.20 × 10−15 | <5 × 10−8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loika, Y.; Loiko, E.; Culminskaya, I.; Kulminski, A.M. Exome-Wide Association Study Identified Clusters of Pleiotropic Genetic Associations with Alzheimer’s Disease and Thirteen Cardiovascular Traits. Genes 2023, 14, 1834. https://doi.org/10.3390/genes14101834
Loika Y, Loiko E, Culminskaya I, Kulminski AM. Exome-Wide Association Study Identified Clusters of Pleiotropic Genetic Associations with Alzheimer’s Disease and Thirteen Cardiovascular Traits. Genes. 2023; 14(10):1834. https://doi.org/10.3390/genes14101834
Chicago/Turabian StyleLoika, Yury, Elena Loiko, Irina Culminskaya, and Alexander M. Kulminski. 2023. "Exome-Wide Association Study Identified Clusters of Pleiotropic Genetic Associations with Alzheimer’s Disease and Thirteen Cardiovascular Traits" Genes 14, no. 10: 1834. https://doi.org/10.3390/genes14101834
APA StyleLoika, Y., Loiko, E., Culminskaya, I., & Kulminski, A. M. (2023). Exome-Wide Association Study Identified Clusters of Pleiotropic Genetic Associations with Alzheimer’s Disease and Thirteen Cardiovascular Traits. Genes, 14(10), 1834. https://doi.org/10.3390/genes14101834