
Complex Genomic Rearrangements Involving ETV6::ABL1 Gene Fusion in an Individual with Myeloid Neoplasm


{kind=link}
Abstract
:1. Introduction
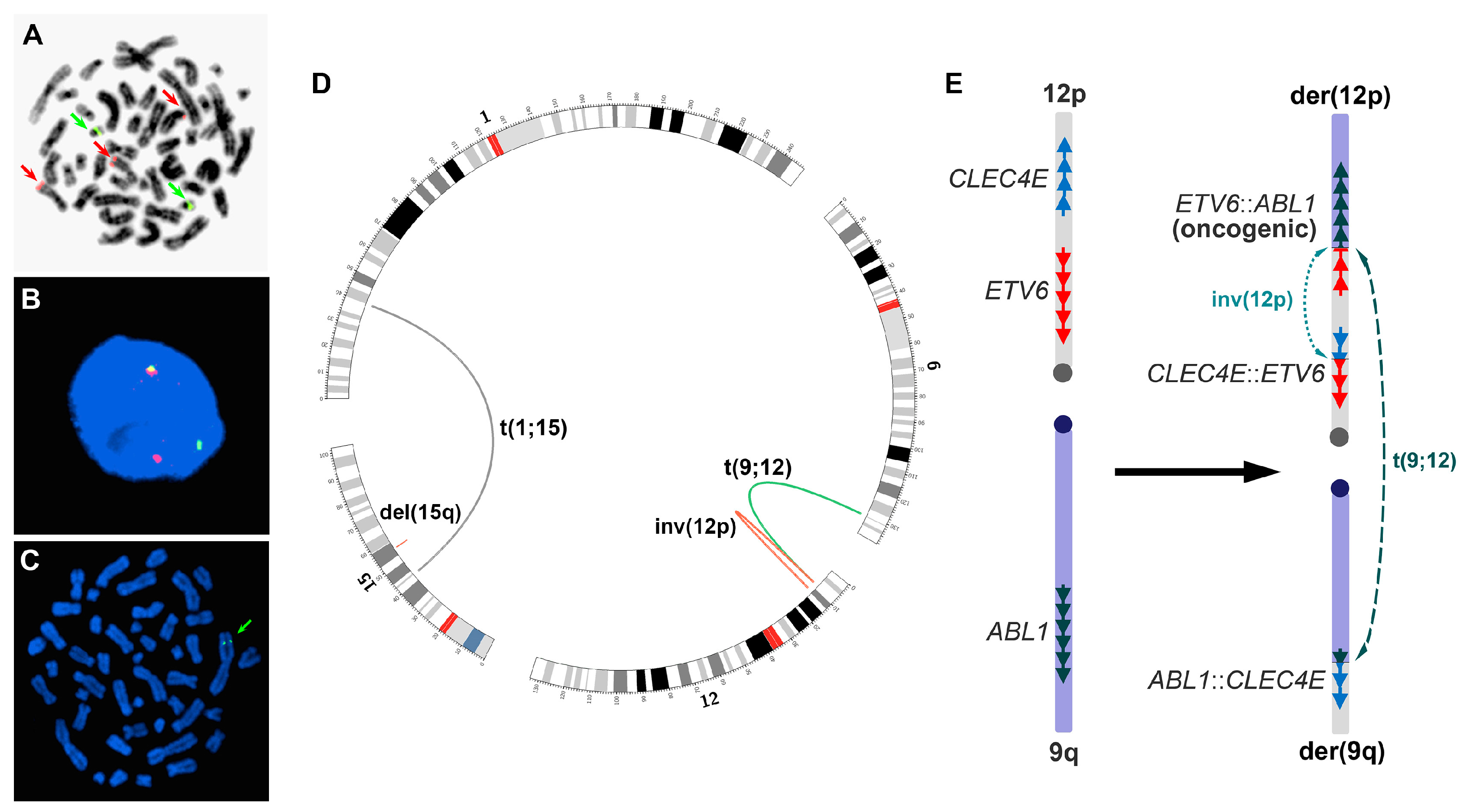
2. Materials and Methods
2.1. Chromosome Analysis
2.2. FISH
2.3. Whole-Genome MP-Seq
2.4. Polymerase Chain Reaction (PCR) and Sanger Sequencing
- TGGGGAGCCCTTATTATTTTT and TTCTATCCCCAAGCCTTCCT for der(1) of t(1;15)(p34;q15),
- CCACATAATCAAAAGTTGACTGC and ACGGCATGAGTCCAGAAGAT for der(15) of t(1;15)(p34;q15),
- ACGCAGCCTTCACTGGTAGT and CTGTCTGCATGTAAACTGTAT for del(15q),
- TTTGTTTTTAGGCAGGCAAA and CAAGACTTTCTGCCCCAATG for der(9) of t(9;12)(q34;p13),
- CAGAGGCAGATAAAAATTCTCCA and GGAAAAGTTTGCCGGATACA for der(12) of t(9;12)(q34;p13),
- CTGGGATCCCCATCCTATT and AGATGAGCAACCCAAGCATC for the proximal breakpoint of inv(12p).
3. Case Presentation
4. Results
4.1. Initial Genomic Testing
4.2. Verifications
4.3. Following-Up Studies
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andreasson, P.; Johansson, B.; Carlsson, M.; Jarlsfelt, I.; Fioretos, T.; Mitelman, F.; Hoglund, M. BCR/ABL-negative chronic myeloid leukemia with ETV6/ABL fusion. Genes Chromosomes Cancer 1997, 20, 299–304. [Google Scholar] [CrossRef]
- La Starza, R.; Trubia, M.; Testoni, N.; Ottaviani, E.; Belloni, E.; Crescenzi, B.; Martelli, M.; Flandrin, G.; Pelicci, P.G.; Mecucci, C. Clonal eosinophils are a morphologic hallmark of ETV6/ABL1 positive acute myeloid leukemia. Haematologica 2002, 87, 789–794. [Google Scholar]
- Uemura, S.; Nishimura, N.; Hasegawa, D.; Shono, A.; Sakaguchi, K.; Matsumoto, H.; Nakamachi, Y.; Saegusa, J.; Yokoi, T.; Tahara, T.; et al. ETV6-ABL1 fusion combined with monosomy 7 in childhood B-precursor acute lymphoblastic leukemia. Int. J. Hematol. 2018, 107, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Yakushijin, K.; Nakamachi, Y.; Miyata, Y.; Sanada, Y.; Tanaka, Y.; Okamura, A.; Kawano, S.; Hayashi, Y.; Matsuoka, H.; et al. Extramedullary T-lymphoid blast crisis of an ETV6/ABL1-positive myeloproliferative neoplasm with t(9;12)(q34;p13) and t(7;14)(p13;q11.2). Ann. Hematol. 2014, 93, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Moorman, A.V.; Cazzaniga, G.; Stanulla, M.; Harvey, R.C.; Roberts, K.G.; Heatley, S.L.; Loh, M.L.; Konopleva, M.; Chen, I.M.; et al. Characterization of leukemias with ETV6-ABL1 fusion. Haematologica 2016, 101, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Xu, L.; Aypar, U.; Meyerson, H.J.; Londono, D.; Gao, Q.; Baik, J.; Dietz, J.; Benayed, R.; Sigler, A.; et al. Myeloid/lymphoid neoplasms with eosinophilia/ basophilia and ETV6-ABL1 fusion: Cell-of-origin and response to tyrosine kinase inhibition. Haematologica 2021, 106, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Schwaab, J.; Naumann, N.; Luebke, J.; Jawhar, M.; Somervaille, T.C.P.; Williams, M.S.; Frewin, R.; Jost, P.J.; Lichtenegger, F.S.; La Rosee, P.; et al. Response to tyrosine kinase inhibitors in myeloid neoplasms associated with PCM1-JAK2, BCR-JAK2 and ETV6-ABL1 fusion genes. Am. J. Hematol. 2020, 95, 824–833. [Google Scholar] [CrossRef]
- Perna, F.; Abdel-Wahab, O.; Levine, R.L.; Jhanwar, S.C.; Imada, K.; Nimer, S.D. ETV6-ABL1-positive “chronic myeloid leukemia”: Clinical and molecular response to tyrosine kinase inhibition. Haematologica 2011, 96, 342–343. [Google Scholar] [CrossRef]
- Lin, H.; Guo, J.Q.; Andreeff, M.; Arlinghaus, R.B. Detection of dual TEL-ABL transcripts and a Tel-Abl protein containing phosphotyrosine in a chronic myeloid leukemia patient. Leukemia 2002, 16, 294–297. [Google Scholar] [CrossRef]
- Jordan, M.; Hastings, R.J.; Moore, S. ISCN 2020: An International System for Human Cytogenomic Nomenclature; Karger International: Basel, Switzerland, 2020; Volume 170. [Google Scholar] [CrossRef]
- Van Limbergen, H.; Beverloo, H.B.; van Drunen, E.; Janssens, A.; Hahlen, K.; Poppe, B.; Van Roy, N.; Marynen, P.; De Paepe, A.; Slater, R.; et al. Molecular cytogenetic and clinical findings in ETV6/ABL1-positive leukemia. Genes Chromosomes Cancer 2001, 30, 274–282. [Google Scholar] [CrossRef]
- Kakadia, P.M.; Schmidmaier, R.; Volkl, A.; Schneider, I.; Huk, N.; Schneider, S.; Panzner, G.; Neidel, U.; Fritz, B.; Spiekermann, K.; et al. An ETV6-ABL1 fusion in a patient with chronic myeloproliferative neoplasm: Initial response to Imatinib followed by rapid transformation into ALL. Leuk. Res. Rep. 2016, 6, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Jang, M.A.; Jeong, W.J.; Jeon, B.R.; Lee, Y.W.; Shin, H.B.; Hong, D.S.; Lee, Y.K. A Case of Chronic Myeloid Leukemia With Rare Variant ETV6/ABL1 Rearrangement. Ann. Lab. Med. 2017, 37, 77–80. [Google Scholar] [CrossRef]
- Lukes, J., Jr.; Potuckova, E.; Sramkova, L.; Stary, J.; Starkova, J.; Trka, J.; Votava, F.; Zuna, J.; Zaliova, M. Two novel fusion genes, AIF1L-ETV6 and ABL1-AIF1L, result together with ETV6-ABL1 from a single chromosomal rearrangement in acute lymphoblastic leukemia with prenatal origin. Genes Chromosomes Cancer 2018, 57, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Renzi, S.; Algawahmed, F.; Davidson, S.; Langenberg, K.P.S.; Fuligni, F.; Ali, S.; Anderson, N.; Brunga, L.; Bartram, J.; Abdelhaleem, M.; et al. Myeloproliferative Neoplasm Driven by ETV6-ABL1 in an Adolescent with Recent History of Burkitt Leukemia. Curr. Oncol. 2023, 30, 5946–5952. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, M.; Lim, J.; Kim, Y.; Han, K.; Kim, J.S.; Lee, S.; Kim, H.J.; Min, W.S. Variant of ETV6/ABL1 gene is associated with leukemia phenotype. Acta Haematol. 2013, 129, 78–82. [Google Scholar] [CrossRef]
- Kelly, J.C.; Shahbazi, N.; Scheerle, J.; Jahn, J.; Suchen, S.; Christacos, N.C.; Mowrey, P.N.; Witt, M.H.; Hostetter, A.; Meloni-Ehrig, A.M. Insertion (12;9)(p13;q34q34): A cryptic rearrangement involving ABL1/ETV6 fusion in a patient with Philadelphia-negative chronic myeloid leukemia. Cancer Genet. Cytogenet. 2009, 192, 36–39. [Google Scholar] [CrossRef]
- Baeumler, J.; Szuhai, K.; Falkenburg, J.H.; van Schie, M.L.; Ottmann, O.G.; Nijmeijer, B.A. Establishment and cytogenetic characterization of a human acute lymphoblastic leukemia cell line (ALL-VG) with ETV6/ABL1 rearrangement. Cancer Genet. Cytogenet. 2008, 185, 37–42. [Google Scholar] [CrossRef]
- Gancheva, K.; Virchis, A.; Howard-Reeves, J.; Cross, N.C.; Brazma, D.; Grace, C.; Kotzampaltiris, P.; Partheniou, F.; Nacheva, E. Myeloproliferative neoplasm with ETV6-ABL1 fusion: A case report and literature review. Mol. Cytogenet. 2013, 6, 39. [Google Scholar] [CrossRef]
- Barbouti, A.; Ahlgren, T.; Johansson, B.; Hoglund, M.; Lassen, C.; Turesson, I.; Mitelman, F.; Fioretos, T. Clinical and genetic studies of ETV6/ABL1-positive chronic myeloid leukaemia in blast crisis treated with imatinib mesylate. Br. J. Haematol. 2003, 122, 85–93. [Google Scholar] [CrossRef]
- Tirado, C.A.; Siangchin, K.; Shabsovich, D.S.; Sharifian, M.; Schiller, G. A novel three-way rearrangement involving ETV6 (12p13) and ABL1 (9q34) with an unknown partner on 3p25 resulting in a possible ETV6-ABL1 fusion in a patient with acute myeloid leukemia: A case report and a review of the literature. Biomark. Res. 2016, 4, 16. [Google Scholar] [CrossRef]
- Song, J.S.; Shin, S.Y.; Lee, S.T.; Kim, H.J.; Kim, S.H. A cryptic ETV6/ABL1 rearrangement represents a unique fluorescence in situ hybridization signal pattern in a patient with B acute lymphoblastic leukemia. Ann. Lab. Med. 2014, 34, 475–477. [Google Scholar] [CrossRef]
- Mori, N.; Ohwashi-Miyazaki, M.; Okada, M.; Yoshinaga, K.; Shiseki, M.; Tanaka, J. Translocation (9;12)(q34.1;p13.?3) Resulted in ETV6-ABL1 Fusion in a Patient with Philadelphia Chromosome-Negative Chronic Myelogenous Leukemia. Acta Haematol. 2016, 136, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.A.; Chelbi, S.T.; Guarda, G. Regulatory Factor X 7 and its Potential Link to Lymphoid Cancers. Trends Cancer 2020, 6, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; de la Rosa, J.; Grove, C.S.; Schick, M.; Rad, L.; Baranov, O.; Strong, A.; Pfaus, A.; Friedrich, M.J.; Engleitner, T.; et al. PiggyBac transposon tools for recessive screening identify B-cell lymphoma drivers in mice. Nat. Commun. 2019, 10, 1415. [Google Scholar] [CrossRef] [PubMed]
- Coronel, L.; Riege, K.; Schwab, K.; Forste, S.; Hackes, D.; Semerau, L.; Bernhart, S.H.; Siebert, R.; Hoffmann, S.; Fischer, M. Transcription factor RFX7 governs a tumor suppressor network in response to p53 and stress. Nucleic Acids Res. 2021, 49, 7437–7456. [Google Scholar] [CrossRef]
- Jessen, S.; Gu, B.; Dai, X. Pygopus and the Wnt signaling pathway: A diverse set of connections. BioEssays News Rev. Mol. Cell. Dev. Biol. 2008, 30, 448–456. [Google Scholar] [CrossRef]
- Kramps, T.; Peter, O.; Brunner, E.; Nellen, D.; Froesch, B.; Chatterjee, S.; Murone, M.; Zullig, S.; Basler, K. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear β-catenin-TCF complex. Cell 2002, 109, 47–60. [Google Scholar] [CrossRef]
- Belenkaya, T.Y.; Han, C.; Standley, H.J.; Lin, X.; Houston, D.W.; Heasman, J.; Lin, X. pygopus Encodes a nuclear protein essential for wingless/Wnt signaling. Development 2002, 129, 4089–4101. [Google Scholar] [CrossRef]
- Boase, N.A.; Kumar, S. NEDD4: The founding member of a family of ubiquitin-protein ligases. Gene 2015, 557, 113–122. [Google Scholar] [CrossRef]
- Chen, C.; Matesic, L.E. The Nedd4-like family of E3 ubiquitin ligases and cancer. Cancer Metastasis Rev. 2007, 26, 587–604. [Google Scholar] [CrossRef]
- Steidl, C.; Leimeister, C.; Klamt, B.; Maier, M.; Nanda, I.; Dixon, M.; Clarke, R.; Schmid, M.; Gessler, M. Characterization of the human and mouse HEY1, HEY2, and HEYL genes: Cloning, mapping, and mutation screening of a new bHLH gene family. Genomics 2000, 66, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, R.; Nakamura, H.; Watanabe, Y. Identification of protogenin, a novel immunoglobulin superfamily gene expressed during early chick embryogenesis. Gene Expr. Patterns 2005, 5, 778–785. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, Z.; Smith, C.; Shah, N.P.; Yu, J. Complex Genomic Rearrangements Involving ETV6::ABL1 Gene Fusion in an Individual with Myeloid Neoplasm. Genes 2023, 14, 1851. https://doi.org/10.3390/genes14101851
Qi Z, Smith C, Shah NP, Yu J. Complex Genomic Rearrangements Involving ETV6::ABL1 Gene Fusion in an Individual with Myeloid Neoplasm. Genes. 2023; 14(10):1851. https://doi.org/10.3390/genes14101851
Chicago/Turabian StyleQi, Zhongxia, Catherine Smith, Neil P. Shah, and Jingwei Yu. 2023. "Complex Genomic Rearrangements Involving ETV6::ABL1 Gene Fusion in an Individual with Myeloid Neoplasm" Genes 14, no. 10: 1851. https://doi.org/10.3390/genes14101851