
Exploring Large MAF Transcription Factors: Functions, Pathology, and Mouse Models with Point Mutations


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
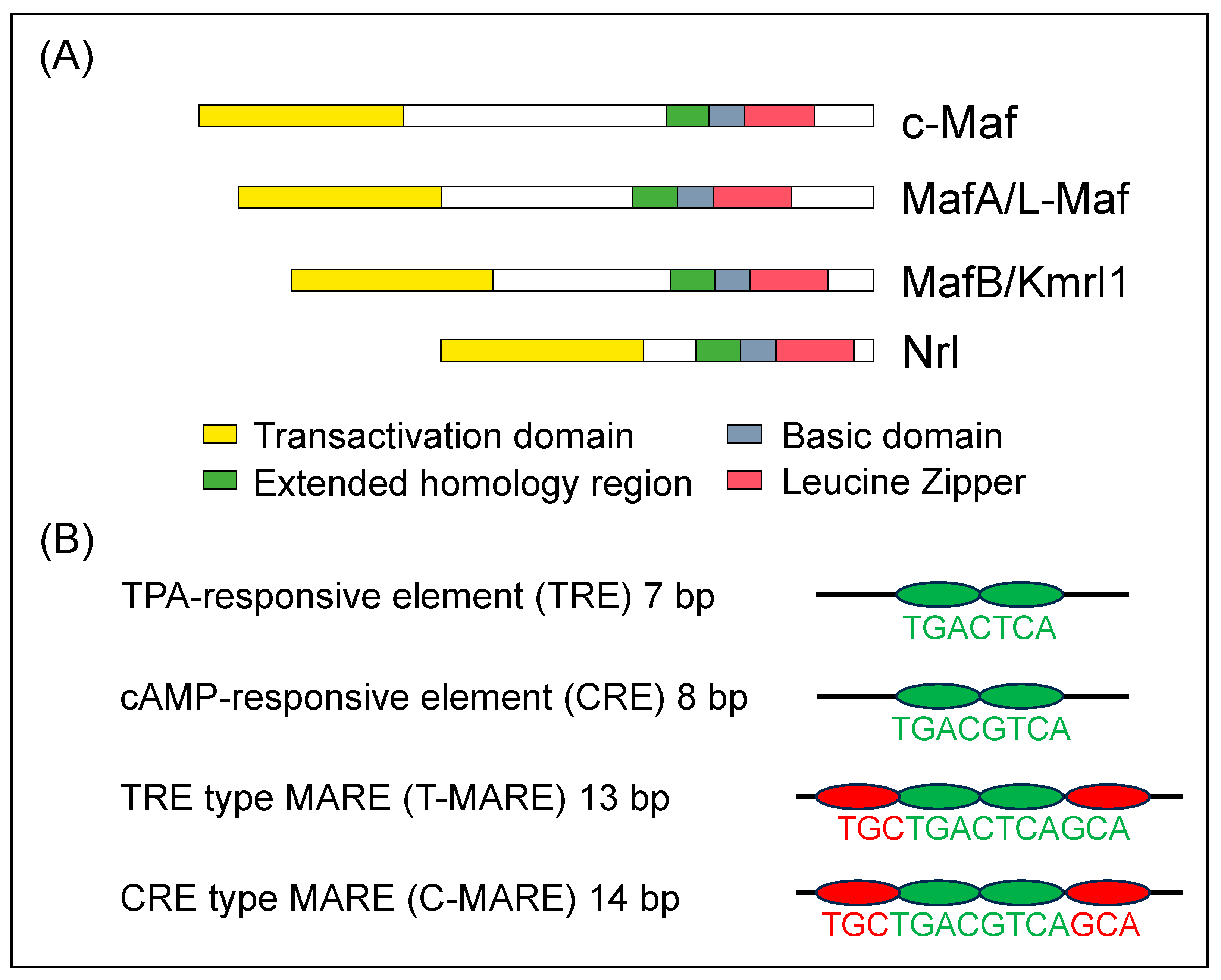
2. Large MAF Transcription Factors
3. Roles of c-MAF
4. c-MAF Point Mutation in Human Patients
5. Model Mouse of c-MAF Human Point Mutation
6. Roles of MAFA
7. MAFA Point Mutation in Human Patients
8. Model Mouse of MafA Human Point Mutation
9. Roles of MAFB
10. MAFB Point Mutation in Human Patients
11. Model Mouse of MAFB Human Point Mutation
12. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishizawa, M.; Kataoka, K.; Goto, N.; Fujiwara, K.T.; Kawai, S. v-maf, a viral oncogene that encodes a “leucine zipper” motif. Proc. Natl. Acad. Sci. USA 1989, 86, 7711–7715. [Google Scholar] [CrossRef]
- Motohashi, H.; Shavit, J.A.; Igarashi, K.; Yamamoto, M.; Engel, J.D. The world according to maf. Nucleic Acids Res. 1997, 25, 2953–2959. [Google Scholar] [CrossRef]
- Blank, V.; Andrews, N.C. The maf transcription factors: Regulators of differentiation. Trends Biochem. Sci. 1997, 22, 437–441. [Google Scholar] [CrossRef]
- Motohashi, H.; Katsuoka, F.; Shavit, J.A.; Engel, J.D.; Yamamoto, M. Positive or negative MARE-dependent transcriptional regulation is determined by the abundance of small maf proteins. Cell 2000, 103, 865–875. [Google Scholar] [CrossRef]
- Yang, Y.; Cvekl, A. Large maf transcription factors: Cousins of AP-1 proteins and important regulators of cellular differentiation. Einstein J. Biol. Med. 2007, 23, 2–11. [Google Scholar] [CrossRef]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef]
- Ho, I.C.; Hodge, M.R.; Rooney, J.W.; Glimcher, L.H. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell 1996, 85, 973–983. [Google Scholar] [CrossRef]
- Blonska, M.; Joo, D.; Nurieva, R.I.; Zhao, X.; Chiao, P.; Sun, S.C.; Dong, C.; Lin, X. Activation of the transcription factor c-Maf in T cells is dependent on the CARMA1-IKKβ signaling cascade. Sci. Signal. 2013, 6, ra110. [Google Scholar] [CrossRef]
- Bauquet, A.T.; Jin, H.; Paterson, A.M.; Mitsdoerffer, M.; Ho, I.C.; Sharpe, A.H.; Kuchroo, V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 2009, 10, 167–175. [Google Scholar] [CrossRef]
- Sato, K.; Miyoshi, F.; Yokota, K.; Araki, Y.; Asanuma, Y.; Akiyama, Y.; Yoh, K.; Takahashi, S.; Aburatani, H.; Mimura, T. Marked induction of c-Maf protein during Th17 cell differentiation and its implication in memory Th cell development. J. Biol. Chem. 2011, 286, 14963–14971. [Google Scholar] [CrossRef]
- Cao, S.; Liu, J.; Song, L.; Ma, X. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J. Immunol. 2005, 174, 3484–3492. [Google Scholar] [CrossRef]
- Kim, J.I.; Li, T.; Ho, I.C.; Grusby, M.J.; Glimcher, L.H. Requirement for the c-Maf transcription factor in crystallin gene regulation and lens development. Proc. Natl. Acad. Sci. USA 1999, 96, 3781–3785. [Google Scholar] [CrossRef]
- Kawauchi, S.; Takahashi, S.; Nakajima, O.; Ogino, H.; Morita, M.; Nishizawa, M.; Yasuda, K.; Yamamoto, M. Regulation of lens fiber cell differentiation by transcription factor c-Maf. J. Biol. Chem. 1999, 274, 19254–19260. [Google Scholar] [CrossRef]
- Liu, F.Y.; Tang, X.C.; Deng, M.; Chen, P.; Ji, W.; Zhang, X.; Gong, L.; Woodward, Z.; Liu, J.; Zhang, L.; et al. The tumor suppressor p53 regulates c-Maf and Prox-1 to control lens differentiation. Curr. Mol. Med. 2012, 12, 917–928. [Google Scholar] [CrossRef]
- Kase, S.; Yoshida, K.; Sakai, M.; Ohgami, K.; Shiratori, K.; Kitaichi, N.; Suzuki, Y.; Harada, T.; Ohno, S. Immunolocalization of cyclin D1 in the developing lens of c-maf -/- mice. Acta Histochem. 2006, 107, 469–472. [Google Scholar] [CrossRef]
- Ring, B.Z.; Cordes, S.P.; Overbeek, P.A.; Barsh, G.S. Regulation of mouse lens fiber cell development and differentiation by the Maf gene. Development 2000, 127, 307–317. [Google Scholar] [CrossRef]
- Nishikawa, K.; Nakashima, T.; Takeda, S.; Isogai, M.; Hamada, M.; Kimura, A.; Kodama, T.; Yamaguchi, A.; Owen, M.J.; Takahashi, S.; et al. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. J. Clin. Investig. 2010, 120, 3455–3465. [Google Scholar] [CrossRef]
- Kusakabe, M.; Hasegawa, K.; Hamada, M.; Nakamura, M.; Ohsumi, T.; Suzuki, H.; Tran, M.T.; Kudo, T.; Uchida, K.; Ninomiya, H.; et al. c-Maf plays a crucial role for the definitive erythropoiesis that accompanies erythroblastic island formation in the fetal liver. Blood 2011, 118, 1374–1385. [Google Scholar] [CrossRef]
- Fujino, M.; Tagami, A.; Ojima, M.; Mizuno, S.; Abdellatif, A.M.; Kuno, A.; Takahashi, S. c-MAF deletion in adult C57BL/6J mice induces cataract formation and abnormal differentiation of lens fiber cells. Exp. Anim. 2020, 69, 242–249. [Google Scholar] [CrossRef]
- Gómez-Salinero, J.M.; Izzo, F.; Lin, Y.; Houghton, S.; Itkin, T.; Geng, F.; Bram, Y.; Adelson, R.P.; Lu, T.M.; Inghirami, G.; et al. Specification of fetal liver endothelial progenitors to functional zonated adult sinusoids requires c-Maf induction. Cell Stem Cell 2022, 29, 593–609.e7. [Google Scholar] [CrossRef]
- González-Loyola, A.; Bernier-Latmani, J.; Roci, I.; Wyss, T.; Langer, J.; Durot, S.; Munoz, O.; Prat-Luri, B.; Delorenzi, M.; Lutolf, M.P.; et al. c-MAF coordinates enterocyte zonation and nutrient uptake transcriptional programs. J. Exp. Med. 2022, 219, e20212418. [Google Scholar] [CrossRef] [PubMed]
- Fujino, M.; Morito, N.; Hayashi, T.; Ojima, M.; Ishibashi, S.; Kuno, A.; Koshiba, S.; Yamagata, K.; Takahashi, S. Transcription factor c-Maf deletion improves streptozotocin-induced diabetic nephropathy by directly regulating Sglt2 and Glut2. JCI Insight 2023, 8, e163306. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, R.V.; Perveen, R.; Kerr, B.; Carette, M.; Yardley, J.; Heon, E.; Wirth, M.G.; van Heyningen, V.; Donnai, D.; Munier, F.; et al. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum. Mol. Genet. 2002, 11, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, R.V.; Munier, F.; Balmer, A.; Farrar, N.; Perveen, R.; Black, G.C. Pulverulent cataract with variably associated microcornea and iris coloboma in a MAF mutation family. Br. J. Ophthalmol. 2003, 87, 411–412. [Google Scholar] [CrossRef]
- Vanita, V.; Singh, D.; Robinson, P.N.; Sperling, K.; Singh, J.R. A novel mutation in the DNA-binding domain of MAF at 16q23.1 associated with autosomal dominant “cerulean cataract” in an Indian family. Am. J. Med. Genet. A 2006, 140, 558–566. [Google Scholar] [CrossRef]
- Wende, H.; Lechner, S.G.; Cheret, C.; Bourane, S.; Kolanczyk, M.E.; Pattyn, A.; Reuter, K.; Munier, F.L.; Carroll, P.; Lewin, G.R.; et al. The transcription factor c-Maf controls touch receptor development and function. Science 2012, 335, 1373–1376. [Google Scholar] [CrossRef] [PubMed]
- Niceta, M.; Stellacci, E.; Gripp, K.W.; Zampino, G.; Kousi, M.; Anselmi, M.; Traversa, A.; Ciolfi, A.; Stabley, D.; Bruselles, A.; et al. Mutations impairing GSK3-mediated MAF phosphorylation cause cataract, deafness, intellectual disability, seizures, and a Down syndrome-like facies. Am. J. Hum. Genet. 2015, 96, 816–825. [Google Scholar] [CrossRef]
- Javadiyan, S.; Craig, J.E.; Sharma, S.; Lower, K.M.; Casey, T.; Haan, E.; Souzeau, E.; Burdon, K.P. Novel missense mutation in the bZIP transcription factor, MAF, associated with congenital cataract, developmental delay, seizures and hearing loss (Aymé-Gripp syndrome). BMC Med. Genet. 2017, 18, 52. [Google Scholar] [CrossRef]
- Niceta, M.; Barbuti, D.; Gupta, N.; Ruggiero, C.; Tizzano, E.F.; Graul-Neumann, L.; Barresi, S.; Nishimura, G.; Valenzuela, I.; López-Grondona, F.; et al. Skeletal abnormalities are common features in Aymé-Gripp syndrome. Clin. Genet. 2020, 97, 362–369. [Google Scholar] [CrossRef]
- Alkhunaizi, E.; Koenekoop, R.K.; Saint-Martin, C.; Russell, L. LMaternally inherited MAF variant associated with variable expression of Aymé-Gripp syndrome. Am. J. Med. Genet. A 2019, 179, 2233–2236. [Google Scholar] [CrossRef]
- König, A.L.; Sabir, H.; Strizek, B.; Gembruch, U.; Herberg, U.; Bertrand, M.; Grasshoff, U.; Wiegand, G.; Wiechers, C.; Bernis, E.; et al. Isolated cytokine-enriched pericardial effusion: A likely key feature for Aymé-Gripp syndrome. Am. J. Med. Genet. A 2022, 188, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Fujino, M.; Ojima, M.; Ishibashi, S.; Mizuno, S.; Takahashi, S. Generation and mutational analysis of a transgenic murine model of the human MAF mutation. Am. J. Med. Genet. A 2023, 191, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- Ogino, H.; Yasuda, K. Induction of lens differentiation by activation of a bZIP transcription factor, L-Maf. Science 1998, 280, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Han, S.I.; Shioda, S.; Hirai, M.; Nishizawa, M.; Handa, H. MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J. Biol. Chem. 2002, 277, 49903–49910. [Google Scholar] [CrossRef]
- Olbrot, M.; Rud, J.; Moss, L.G.; Sharma, A. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc. Natl. Acad. Sci. USA 2002, 99, 6737–6742. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Zhao, L.; Artner, I.; Jarrett, H.W.; Friedman, D.; Means, A.; Stein, R. Members of the large Maf transcription family regulate insulin gene transcription in islet β cells. Mol. Cell. Biol. 2003, 23, 6049–6062. [Google Scholar] [CrossRef]
- Kataoka, K.; Shioda, S.; Ando, K.; Sakagami, K.; Handa, H.; Yasuda, K. Differentially expressed Maf family transcription factors, c-Maf and MafA, activate glucagon and insulin gene expression in pancreatic islet α- and β-cells. J. Mol. Endocrinol. 2004, 32, 9–20. [Google Scholar] [CrossRef]
- Artner, I.; Le Lay, J.; Hang, Y.; Elghazi, L.; Schisler, J.C.; Henderson, E.; Sosa-Pineda, B.; Stein, R. MafB: An activator of the glucagon gene expressed in developing islet α- and β-cells. Diabetes 2006, 55, 297–304. [Google Scholar] [CrossRef]
- Artner, I.; Blanchi, B.; Raum, J.C.; Guo, M.; Kaneko, T.; Cordes, S.; Sieweke, M.; Stein, R. MafB is required for islet β cell maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 3853–3858. [Google Scholar] [CrossRef]
- Hang, Y.; Stein, R. MafA and MafB activity in pancreatic β cells. Trends Endocrinol. Metab. 2011, 22, 364–373. [Google Scholar] [CrossRef]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005, 25, 4969–4976. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Shimano, H.; Yamamoto, T.; Yokoo, T.; Endo, Y.; Ishikawa, M.; Matsuzaka, T.; Nakagawa, Y.; Kumadaki, S.; Yahagi, N.; et al. Granuphilin is activated by SREBP-1c and involved in impaired insulin secretion in diabetic mice. Cell Metab. 2006, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Iacovazzo, D.; Flanagan, S.E.; Walker, E.; Quezado, R.; de Sousa Barros, F.A.; Caswell, R.; Johnson, M.B.; Wakeling, M.; Brändle, M.; Guo, M.; et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc. Natl. Acad. Sci. USA 2018, 115, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.M.; Cha, J.; Tong, X.; Guo, M.; Liu, J.H.; Yu, S.; Iacovazzo, D.; Mauvais-Jarvis, F.; Flanagan, S.E.; Korbonits, M.; et al. Sex-biased islet β cell dysfunction is caused by the MODY MAFA S64F variant by inducing premature aging and senescence in males. Cell Rep. 2021, 37, 109813. [Google Scholar] [CrossRef] [PubMed]
- Cordes, S.P.; Barsh, G.S. The mouse segmentation gene kr encodes a novel basic domain-leucine zipper transcription factor. Cell 1994, 79, 1025–1034. [Google Scholar] [CrossRef]
- Kataoka, K.; Fujiwara, K.T.; Noda, M.; Nishizawa, M. MafB, a new maf family transcription activator that can associate with maf and Fos but not with Jun. Mol. Cell. Biol. 1994, 14, 7581–7591. [Google Scholar] [CrossRef]
- Sieweke, M.H.; Tekotte, H.; Frampton, J.; Graf, T. MafB is an interaction partner and repressor of Ets-1 that inhibits erythroid differentiation. Cell 1996, 85, 49–60. [Google Scholar] [CrossRef]
- Kelly, L.M.; Englmeier, U.; Lafon, I.; Sieweke, M.H.; Graf, T. MafB is an inducer of monocytic differentiation. EMBO J. 2000, 19, 1987–1997. [Google Scholar] [CrossRef]
- Sadl, V.; Jin, F.; Yu, J.; Cui, S.; Holmyard, D.; Quaggin, S.; Barsh, G.; Cordes, S. The mouse Kreisler (Krml1/MafB) segmentation gene is required for differentiation of glomerular visceral epithelial cells. Dev. Biol. 2002, 249, 16–29. [Google Scholar] [CrossRef]
- Blanchi, B.; Kelly, L.M.; Viemari, J.C.; Lafon, I.; Burnet, H.; Bévengut, M.; Tillmanns, S.; Daniel, L.; Graf, T.; Hilaire, G.; et al. MafB deficiency causes defective respiratory rhythmogenesis and fatal central apnea at birth. Nat. Neurosci. 2003, 6, 1091–1100. [Google Scholar] [CrossRef]
- Moriguchi, T.; Hamada, M.; Morito, N.; Terunuma, T.; Hasegawa, K.; Zhang, C.; Yokomizo, T.; Esaki, R.; Kuroda, E.; Yoh, K.; et al. MafB is essential for renal development and F4/80 expression in macrophages. Mol. Cell. Biol. 2006, 26, 5715–5727. [Google Scholar] [CrossRef]
- Aziz, A.; Vanhille, L.; Mohideen, P.; Kelly, L.M.; Otto, C.; Bakri, Y.; Mossadegh, N.; Sarrazin, S.; Sieweke, M.H. Development of macrophages with altered actin organization in the absence of MafB. Mol. Cell. Biol. 2006, 26, 6808–6818. [Google Scholar] [CrossRef] [PubMed]
- Kamitani-Kawamoto, A.; Hamada, M.; Moriguchi, T.; Miyai, M.; Saji, F.; Hatamura, I.; Nishikawa, K.; Takayanagi, H.; Hitoshi, S.; Ikenaka, K.; et al. MafB interacts with Gcm2 and regulates parathyroid hormone expression and parathyroid development. J. Bone Miner. Res. 2011, 26, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Mossadegh-Keller, N.; Fukao, T.; Aziz, A.; Mourcin, F.; Vanhille, L.; Kelly Modis, L.; Kastner, P.; Chan, S.; Duprez, E.; et al. MafB restricts M-CSF-dependent myeloid commitment divisions of hematopoietic stem cells. Cell 2009, 138, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.; Soucie, E.; Sarrazin, S.; Sieweke, M.H. MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages. Science 2009, 326, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Sultana, D.A.; Tomita, S.; Hamada, M.; Iwanaga, Y.; Kitahama, Y.; Khang, N.V.; Hirai, S.; Ohigashi, I.; Nitta, S.; Amagai, T.; et al. Gene expression profile of the third pharyngeal pouch reveals role of mesenchymal MafB in embryonic thymus development. Blood 2009, 113, 2976–2987. [Google Scholar] [CrossRef]
- Miyai, M.; Tanaka, Y.G.; Kamitani, A.; Hamada, M.; Takahashi, S.; Kataoka, K. c-Maf and MafB transcription factors are differentially expressed in Huxley’s and Henle’s layers of the inner root sheath of the hair follicle and regulate cuticle formation. J. Dermatol. Sci. 2010, 57, 178–182. [Google Scholar] [CrossRef]
- Miyai, M.; Hamada, M.; Moriguchi, T.; Hiruma, J.; Kamitani-Kawamoto, A.; Watanabe, H.; Hara-Chikuma, M.; Takahashi, K.; Takahashi, S.; Kataoka, K. Transcription factor MafB coordinates epidermal keratinocyte differentiation. J. Investig. Dermatol. 2016, 136, 1848–1857. [Google Scholar] [CrossRef]
- Suzuki, K.; Numata, T.; Suzuki, H.; Raga, D.D.; Ipulan, L.A.; Yokoyama, C.; Matsushita, S.; Hamada, M.; Nakagata, N.; Nishinakamura, R.; et al. Sexually dimorphic expression of Mafb regulates masculinization of the embryonic urethral formation. Proc. Natl. Acad. Sci. USA 2014, 111, 16407–16412. [Google Scholar] [CrossRef]
- Dieterich, L.C.; Klein, S.; Mathelier, A.; Sliwa-Primorac, A.; Ma, Q.; Hong, Y.K.; Shin, J.W.; Hamada, M.; Lizio, M.; Itoh, M.; et al. DeepCAGE transcriptomics reveal an important role of the transcription factor MAFB in the lymphatic endothelium. Cell Rep. 2015, 13, 1493–1504. [Google Scholar] [CrossRef]
- Hamada, M.; Nakamura, M.; Tran, M.T.; Moriguchi, T.; Hong, C.; Ohsumi, T.; Dinh, T.T.; Kusakabe, M.; Hattori, M.; Katsumata, T.; et al. MafB promotes atherosclerosis by inhibiting foam-cell apoptosis. Nat. Commun. 2014, 5, 3147. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.T.N.; Hamada, M.; Jeon, H.; Shiraishi, R.; Asano, K.; Hattori, M.; Nakamura, M.; Imamura, Y.; Tsunakawa, Y.; Fujii, R.; et al. MafB is a critical regulator of complement component C1q. Nat. Commun. 2017, 8, 1700. [Google Scholar] [CrossRef] [PubMed]
- Shichita, T.; Ito, M.; Morita, R.; Komai, K.; Noguchi, Y.; Ooboshi, H.; Koshida, R.; Takahashi, S.; Kodama, T.; Yoshimura, A. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat. Med. 2017, 23, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Morito, N.; Shawki, H.H.; Sato, Y.; Tsukaguchi, H.; Hamada, M.; Jeon, H.; Yadav, M.K.; Kuno, A.; Tsunakawa, Y.; et al. Transcription factor MafB in podocytes protects against the development of focal segmental glomerulosclerosis. Kidney Int. 2020, 98, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.C.; Jung, Y.; Ugboma, C.M.; Shimbo, M.; Kuno, A.; Basha, W.A.; Kudo, T.; Oishi, H.; Takahashi, S. MafB is critical for glucagon production and secretion in mouse pancreatic α cells in vivo. Mol. Cell. Biol. 2018, 38, e00504-17. [Google Scholar] [CrossRef]
- Xiafukaiti, G.; Maimaiti, S.; Ogata, K.; Kuno, A.; Kudo, T.; Shawki, H.H.; Oishi, H.; Takahashi, S. MafB is important for pancreatic β-cell maintenance under a MafA-deficient condition. Mol. Cell. Biol. 2019, 39, e00080-19. [Google Scholar] [CrossRef]
- Zankl, A.; Duncan, E.L.; Leo, P.J.; Clark, G.R.; Glazov, E.A.; Addor, M.C.; Herlin, T.; Kim, C.A.; Leheup, B.P.; McGill, J.; et al. Multicentric carpotarsal osteolysis is caused by mutations clustering in the amino-terminal transcriptional activation domain of MAFB. Am. J. Hum. Genet. 2012, 90, 494–501. [Google Scholar] [CrossRef]
- Park, J.G.; Tischfield, M.A.; Nugent, A.A.; Cheng, L.; Di Gioia, S.A.; Chan, W.M.; Maconachie, G.; Bosley, T.M.; Summers, C.G.; Hunter, D.G.; et al. Loss of MAFB function in humans and mice causes Duane syndrome, aberrant extraocular muscle innervation, and inner-ear defects. Am. J. Hum. Genet. 2016, 98, 1220–1227. [Google Scholar] [CrossRef]
- Sato, Y.; Tsukaguchi, H.; Morita, H.; Higasa, K.; Tran, M.T.N.; Hamada, M.; Usui, T.; Morito, N.; Horita, S.; Hayashi, T.; et al. A mutation in transcription factor MAFB causes Focal Segmental glomerulosclerosis with Duane retraction syndrome. Kidney Int. 2018, 94, 396–407. [Google Scholar] [CrossRef]
- Tsunakawa, Y.; Hamada, M.; Matsunaga, Y.; Fuseya, S.; Jeon, H.; Wakimoto, Y.; Usui, T.; Kanai, M.; Mizuno, S.; Morito, N.; et al. Mice harboring an MCTO mutation exhibit renal failure resembling nephropathy in human patients. Exp. Anim. 2019, 68, 103–111. [Google Scholar] [CrossRef]
- Mehawej, C.; Courcet, J.B.; Baujat, G.; Mouy, R.; Gérard, M.; Landru, I.; Gosselin, M.; Koehrer, P.; Mousson, C.; Breton, S.; et al. The identification of MAFB mutations in eight patients with multicentric carpo-tarsal osteolysis supports genetic homogeneity but clinical variability. Am. J. Med. Genet. A 2013, 161, 3023–3029. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Jeon, H.; Ojima, M.; Nishino, T.; Usui, T.; Yadav, M.K.; Kulathunga, K.; Morito, N.; Takahashi, S.; Hamada, M. Phenotypic analysis of mice carrying human-type MAFB p.Leu239Pro mutation. Biochem. Biophys. Res. Commun. 2020, 523, 452–457. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujino, M.; Ojima, M.; Takahashi, S. Exploring Large MAF Transcription Factors: Functions, Pathology, and Mouse Models with Point Mutations. Genes 2023, 14, 1883. https://doi.org/10.3390/genes14101883
Fujino M, Ojima M, Takahashi S. Exploring Large MAF Transcription Factors: Functions, Pathology, and Mouse Models with Point Mutations. Genes. 2023; 14(10):1883. https://doi.org/10.3390/genes14101883
Chicago/Turabian StyleFujino, Mitsunori, Masami Ojima, and Satoru Takahashi. 2023. "Exploring Large MAF Transcription Factors: Functions, Pathology, and Mouse Models with Point Mutations" Genes 14, no. 10: 1883. https://doi.org/10.3390/genes14101883