
The Genome of the Korean Island-Originated Perilla citriodora ‘Jeju17’ Sheds Light on Its Environmental Adaptation and Fatty Acid and Lipid Production Pathways

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Whole-Genome Sequencing, Genome Size Estimation, and Genome Assembly
2.2. Genome Assembly Quality Assessment
2.3. Repeat Identification and Genome Annotation
2.4. Gene Family Analysis, Time Divergence Estimation, and Phylogenetic Placement of Jeju 17
2.5. Identification of Putative Orthologs
2.6. Identification of Genes Involved in Fatty Acid and Lipid Triacylglycerol Biosynthesis
2.7. Synteny Analysis
3. Results and Discussion
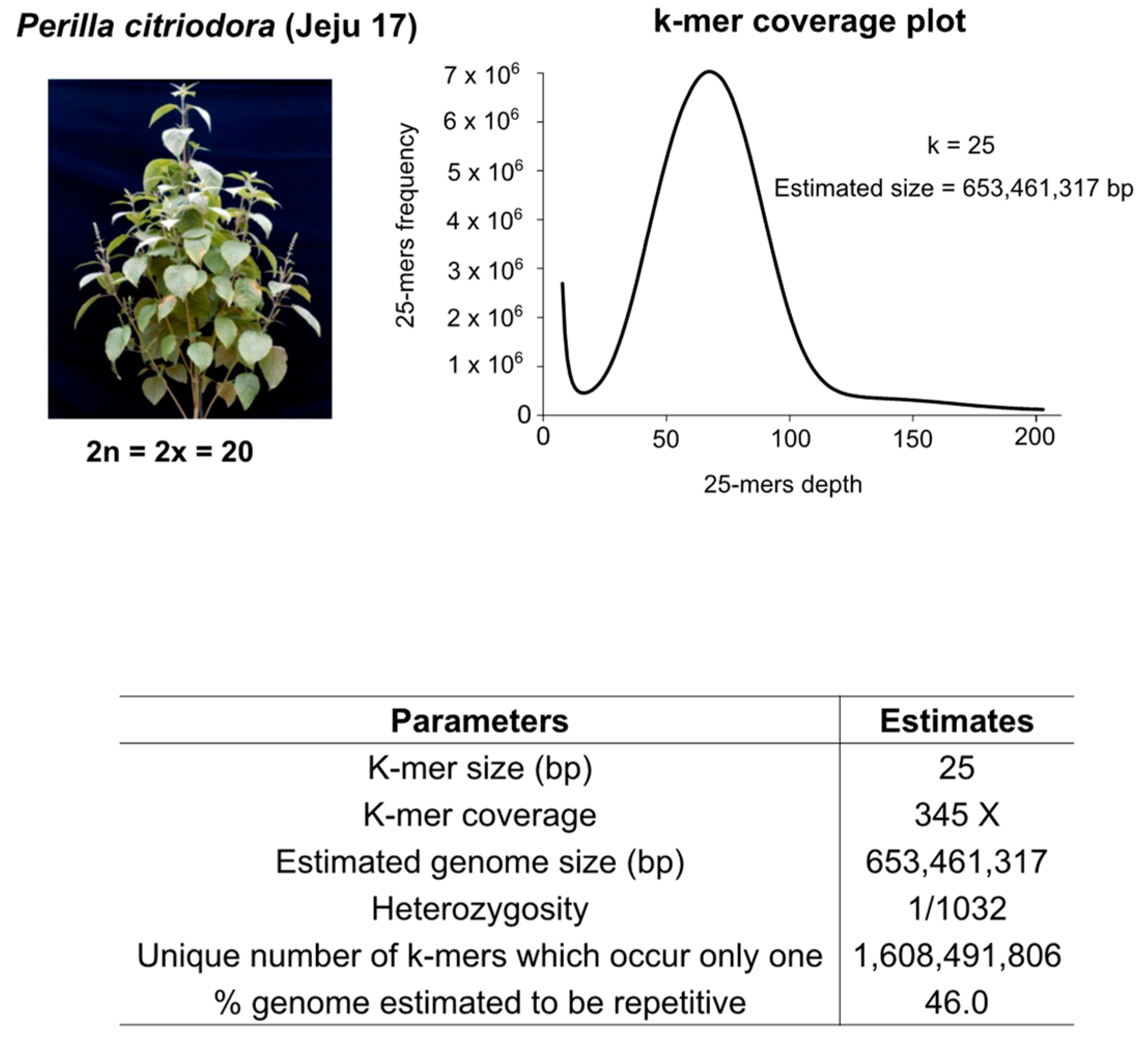
3.1. Genome Survey, Sequencing, and Assembly Quality
3.2. Genome Annotation Features
3.3. Genome Evolution
3.4. Phylogenetic Placement of ‘Jeju17’-Specific Gene Family Assessment
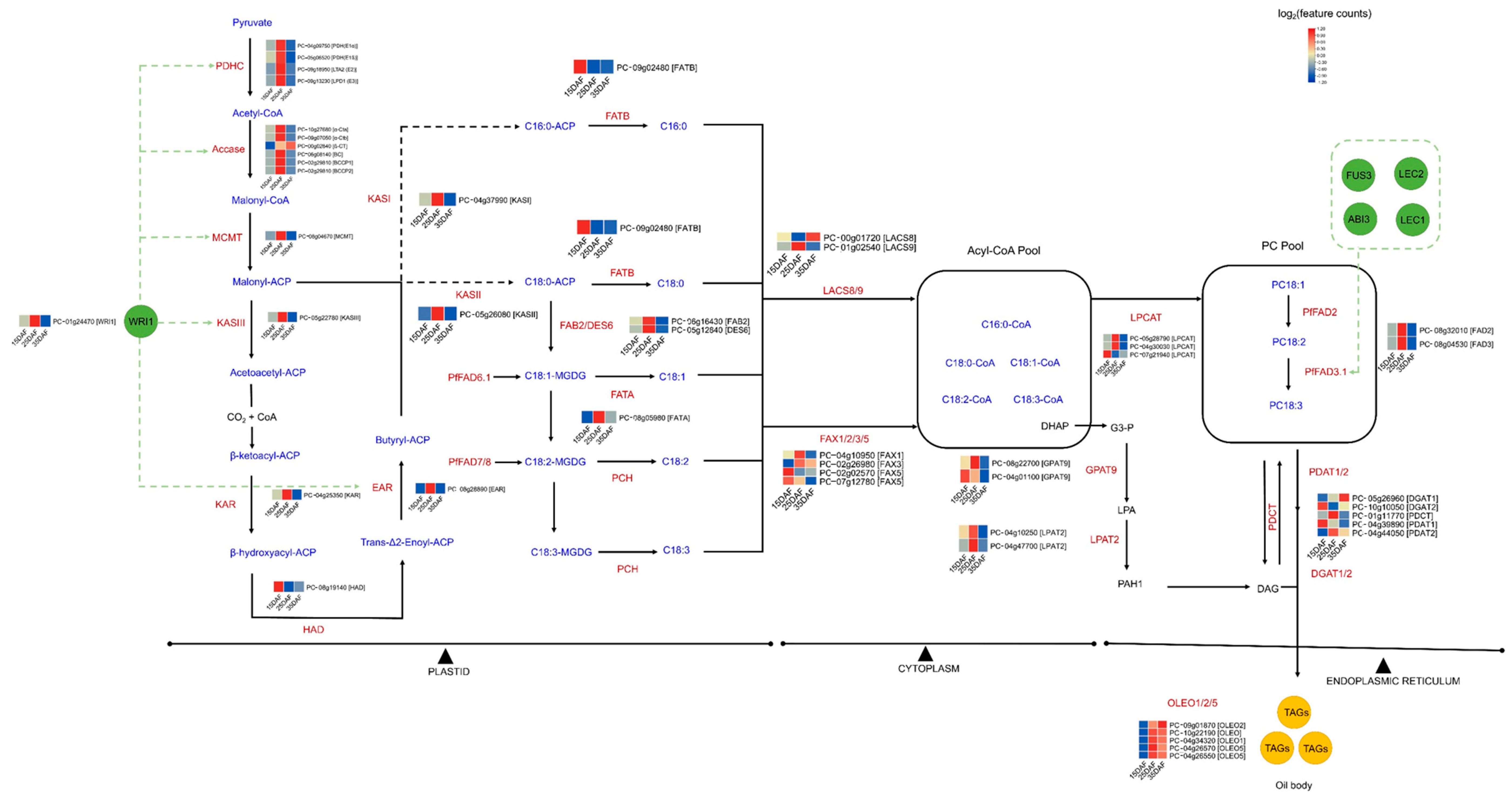
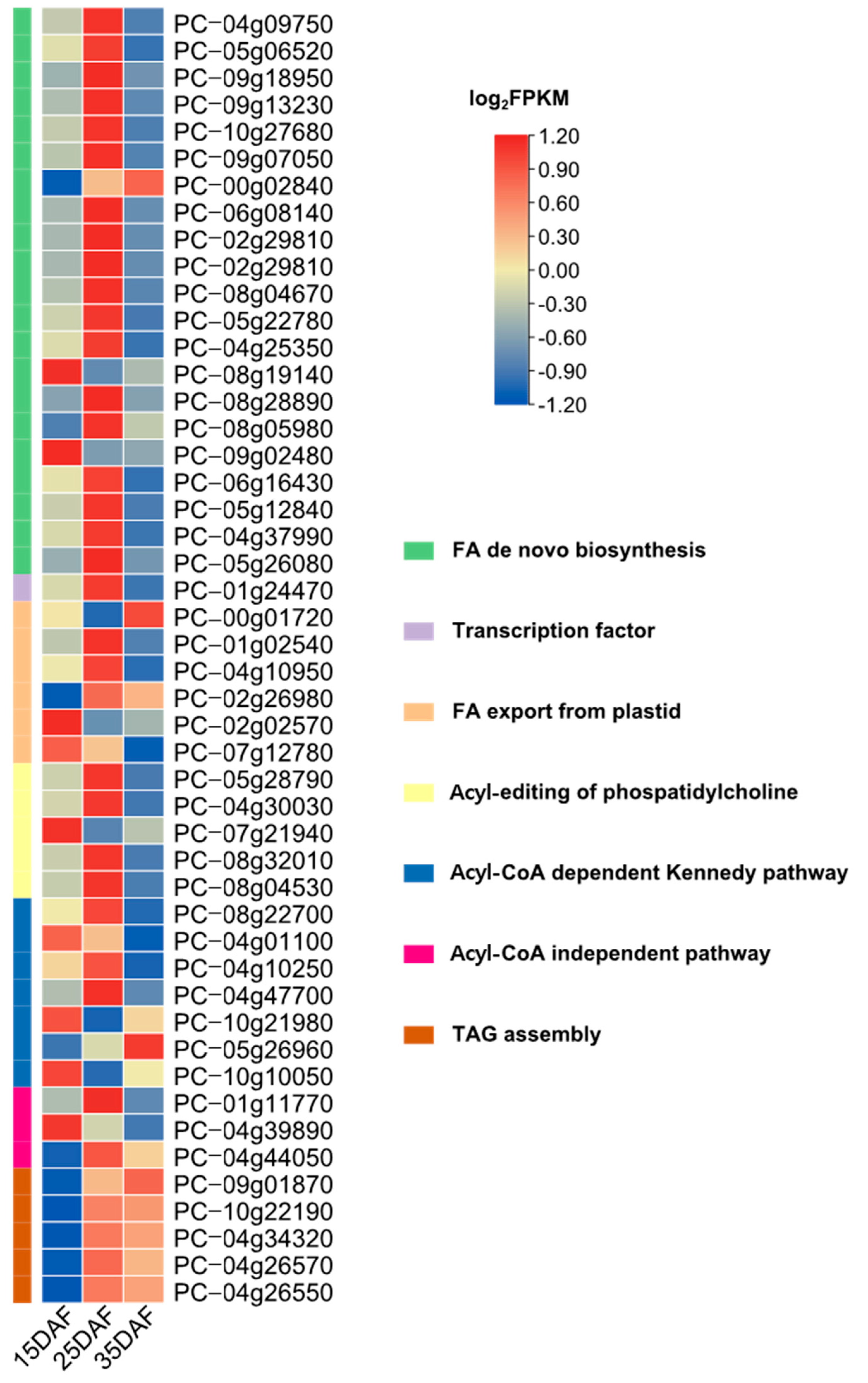
3.5. Identification of Genes Involved in Fatty Acid and Lipid Triacylglycerol Biosynthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nitta, M.; Lee, J.K.; Kang, C.W.; Katsuta, M.; Yasumoto, S.; Liu, D.; Nagamine, T.; Ohnishi, O. The Distribution of Perilla Species. Genet. Resour. Crop Evol. 2005, 52, 797–804. [Google Scholar] [CrossRef]
- Nitta, M.; Lee, J.K.; Ohnishi, O. Asian Perilla crops and their weedy forms: Their cultivation, utilization and genetic relationships. Econ. Bot. 2003, 57, 245–253. [Google Scholar] [CrossRef]
- Cui, X.; Gou, Z.; Fan, Q.; Li, L.; Lin, X.; Wang, Y.; Jiang, S.; Jiang, Z. Effects of dietary perilla seed oil supplementation on lipid metabolism, meat quality, and fatty acid profiles in Yellow-feathered chickens. Poult. Sci. 2019, 98, 5714–5723. [Google Scholar] [CrossRef] [PubMed]
- Arjin, C.; Souphannavong, C.; Norkeaw, R.; Chaiwang, N.; Mekchay, S.; Sartsook, A.; Thongkham, M.; Yosen, T.; Ruksiriwanich, W.; Sommano, S.R.; et al. Effects of Dietary Perilla Cake Supplementation in Growing Pig on Productive Performance, Meat Quality, and Fatty Acid Profiles. Animals 2021, 11, 3213. [Google Scholar] [CrossRef]
- Peiretti, P.G.; Gasco, L.; Brugiapaglia, A.; Gai, F. Effects of perilla (Perilla frutescens L.) seeds supplementation on performance, carcass characteristics, meat quality and fatty acid composition of rabbits. Livest. Sci. 2011, 138, 118–124. [Google Scholar] [CrossRef]
- Xia, J.Q.; He, X.; Wang, L.; Wang, L.; Zhang, D.J.; Wang, J.F.; Liu, D. Evaluation of dietary Perilla frutescens seed on performance and carcass quality in finishing castrated male Songliao black pigs. Vet. Med. Sci. 2022, 8, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shen, Q.; Leng, L.; Zhang, D.; Chen, S.; Shi, Y.; Ning, Z.; Chen, S. Incipient diploidization of the medicinal plant Perilla within 10,000 years. Nat. Commun. 2021, 12, 5508. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Qiu, J.-F.; Ma, L.-J.; Hu, Y.-J.; Li, P.; Wan, J.-B. Phytochemical and phytopharmacological review of Perilla frutescens L. (Labiatae), a traditional edible-medicinal herb in China. Food Chem. Toxicol. 2017, 108, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Netala, V.R.; Zhang, H.; Xing, Y.; Li, H.; Zhang, Z. Perilla frutescens: A Rich Source of Pharmacological Active Compounds. Molecules 2022, 27, 3578. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-S.; Kuo, C.-L.; Wang, J.-P.; Cheng, J.-S.; Huang, Z.-W.; Chen, C.-F. Growth inhibitory and apoptosis inducing effect of Perilla frutescens extract on human hepatoma HepG2 cells. J. Ethnopharmacol. 2007, 112, 557–567. [Google Scholar] [CrossRef]
- Narisawa, T.; Takahashi, M.; Kotanagi, H.; Kusaka, H.; Yamazaki, Y.; Koyama, H.; Fukaura, Y.; Nishizawa, Y.; Kotsugai, M.; Isoda, Y.; et al. Inhibitory Effect of Dietary Perilla Oil Rich in the n-3 Polyunsaturated Fatty Acid α-Linolenic Acid on Colon Carcinogenesis in Rats. Jpn. J. Cancer Res. 1991, 82, 1089–1096. [Google Scholar] [CrossRef]
- Banno, N.; Akihisa, T.; Tokuda, H.; Yasukawa, K.; Higashihara, H.; Ukiya, M.; Watanabe, K.; Kimura, Y.; Hasegawa, J.I.; Nishino, H. Triterpene acids from the leaves of Perilla frutescens and their anti-inflammatory and antitumor-promoting effects. Biosci. Biotechnol. Biochem. 2004, 68, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Leu, Y.-L.; Fang, Y.; Lin, C.-F.; Kuo, L.-M.; Sung, W.-C.; Tsai, Y.-F.; Chung, P.-J.; Lee, M.-C.; Kuo, Y.-T.; et al. Anti-inflammatory effects of Perilla frutescens in activated human neutrophils through two independent pathways: Src family kinases and Calcium. Sci. Rep. 2016, 5, 18204. [Google Scholar] [CrossRef]
- Jeon, I.; Kim, H.; Kang, H.; Lee, H.-S.; Jeong, S.; Kim, S.; Jang, S. Anti-Inflammatory and Antipruritic Effects of Luteolin from Perilla (P. frutescens L.) Leaves. Molecules 2014, 19, 6941–6951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tian, Y.; Guan, J.; Xie, Q.; Zhao, Y. The anti-tussive, anti-inflammatory effects and sub-chronic toxicological evaluation of perilla seed oil. J. Sci. Food Agric. 2021, 101, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Wijendran, V.; Hayes, K.C. Dietary n-6 and n-3 fatty acid balance and cardiovascular health. Annu. Rev. Nutr. 2004, 24, 597–615. [Google Scholar] [CrossRef]
- Wang, F.; Zhu, H.; Hu, M.; Wang, J.; Xia, H.; Yang, X.; Yang, L.; Sun, G. Perilla Oil Supplementation Improves Hypertriglyceridemia and Gut Dysbiosis in Diabetic KKAy Mice. Mol. Nutr. Food Res. 2018, 62, 1800299. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, S.; Li, W.; Ma, L.; Ding, M.; Li, R.; Liu, Y. High-fat diet from perilla oil induces insulin resistance despite lower serum lipids and increases hepatic fatty acid oxidation in rats. Lipids Health Dis. 2014, 13, 15. [Google Scholar] [CrossRef]
- Song, Y.; Sun, R.; Ji, Z.; Li, X.; Fu, Q.; Ma, S. Perilla aldehyde attenuates CUMS-induced depressive-like behaviors via regulating TXNIP/TRX/NLRP3 pathway in rats. Life Sci. 2018, 206, 117–124. [Google Scholar] [CrossRef]
- Nakazawa, T.; Yasuda, T.; Ueda, J.; Ohsawa, K. Antidepressant-like effects of apigenin and 2,4,5-trimethoxycinnamic acid from Perilla frutescens in the forced swimming test. Biol. Pharm. Bull. 2003, 26, 474–480. [Google Scholar] [CrossRef]
- Takeda, H.; Tsuji, M.; Inazu, M.; Egashira, T.; Matsumiya, T. Rosmarinic acid and caffeic acid produce antidepressive-like effect in the forced swimming test in mice. Eur. J. Pharmacol. 2002, 449, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Tsuji, M.; Miyamoto, J.; Matsumiya, T. Rosmarinic acid and caffeic acid reduce the defensive freezing behavior of mice exposed to conditioned fear stress. Psychopharmacology 2002, 164, 233–235. [Google Scholar] [CrossRef]
- Makino, T.; Furuta, Y.; Wakushima, H.; Fujii, H.; Saito, K.; Kano, Y. Anti-allergic effect of Perilla frutescens and its active constituents. Phyther. Res. 2003, 17, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.-L.; Yu, S.-J.; Huang, W.-C.; Yen, H.-R. Luteolin Attenuates Allergic Nasal Inflammation via Inhibition of Interleukin-4 in an Allergic Rhinitis Mouse Model and Peripheral Blood from Human Subjects with Allergic Rhinitis. Front. Pharmacol. 2020, 11, 291. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, T.; Fukaura, Y.; Ishikawa, C.; Isoda, Y.; Nishizawa, N. Colon cancer prevention with a small amount of dietary perilla oil high in alpha-linolenic acid in an animal model. Cancer 1994, 73, 2069–2075. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Noh, S. Positional distribution of fatty acids in perilla (Perilla frutescens L.) oil. J. Am. Oil Chem. Soc. 2011, 88, 157–158. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, B.; Win, A.; Fu, C.; Lian, J.; Liu, X.; Wang, R.; Zhang, X.; Chai, Y. Omega-3 fatty acid desaturase gene family from two ω-3 sources, Salvia hispanica and Perilla frutescens: Cloning, characterization and expression. PLoS ONE 2018, 13, e0191432. [Google Scholar] [CrossRef]
- Zhu, S.; Zhu, Z.; Wang, H.; Wang, L.; Cheng, L.; Yuan, Y.; Li, D. Characterization and functional analysis of a plastidial FAD6 gene and its promoter in the mesocarp of oil palm (Elaeis guineensis). Sci. Hortic. 2018, 239, 163–170. [Google Scholar] [CrossRef]
- Lee, K.-R.; Lee, Y.; Kim, E.-H.; Lee, S.-B.; Roh, K.-H.; Kim, J.-B.; Kim, H.-U. Functional identification of oleate 12-desaturase and ω-3 fatty acid desaturase genes from Perilla frutescens var. frutescens. Plant Cell Rep. 2016, 35, 2523–2537. [Google Scholar] [CrossRef]
- Bae, S.-H.; Zoclanclounon, Y.A.B.; Kumar, T.S.; Oh, J.-H.; Lee, J.; Kim, T.-H.; Park, K.Y. Advances in Understanding the Genetic Basis of Fatty Acids Biosynthesis in Perilla: An Update. Plants 2022, 11, 1207. [Google Scholar] [CrossRef]
- Jung, C.; Lee, M.; Oh, K.; Kim, H.; Park, C.; Sung, J.; Suh, D. Discovery of New Diploid Perilla Species in Korea. Korean J. Breed 2005, 37, 2003–2005. [Google Scholar]
- Sohn, Y.K.; Park, K.H. Early-stage volcanism and sedimentation of Jeju Island revealed by the Sagye borehole, SW Jeju Island, Korea. Geosci. J. 2004, 8, 73–84. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H.; et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the Gap: Upgrading Genomes with Pacific Biosciences RS Long-Read Sequencing Technology. PLoS ONE 2012, 7, e47768. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef]
- Huang, S.; Kang, M.; Xu, A. HaploMerger2: Rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 2017, 33, 2577–2579. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Jain, C.; Koren, S.; Dilthey, A.; Phillippy, A.M.; Aluru, S. A fast adaptive algorithm for computing whole-genome homology maps. Bioinformatics 2018, 34, i748–i756. [Google Scholar] [CrossRef] [PubMed]
- Cabanettes, F.; Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Nattestad, M.; Schatz, M.C. Assemblytics: A web analytics tool for the detection of variants from an assembly. Bioinformatics 2016, 32, 3021–3023. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-H.; Pandey, R.P.; Lee, C.-M.; Sim, J.-S.; Jeong, J.-T.; Choi, B.-S.; Jung, M.; Ginzburg, D.; Zhao, K.; Won, S.Y.; et al. Genome-enabled discovery of anthraquinone biosynthesis in Senna tora. Nat. Commun. 2020, 11, 5875. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Chen, G.Q. Identification of hydroxy fatty acid and triacylglycerol metabolism-related genes in lesquerella through seed transcriptome analysis. BMC Genom. 2015, 16, 230. [Google Scholar] [CrossRef]
- Kaczorowski, T.; Szybalski, W. Genomic DNA sequencing by SPEL-6 primer walking using hexamer ligation. Gene 1998, 223, 83–91. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/2010 (accessed on 7 December 2016).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Mello, B. Estimating TimeTrees with MEGA and the TimeTree Resource. Mol. Biol. Evol. 2018, 35, 2334–2342. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Tao, Q.; Kumar, S. Theoretical Foundation of the RelTime Method for Estimating Divergence Times from Variable Evolutionary Rates. Mol. Biol. Evol. 2018, 35, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Wang, X.; Lee, T.-H.; Jakob, K.; Lee, G.-J.; Paterson, A.H. Comparative analysis of Miscanthus and Saccharum reveals a shared whole genome duplication but different evolutionary fates. Plant Cell 2014, 26, 2420–2429. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Higgins, D.G. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon align ments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Koo, A.J.K.; Ruuska, S.; Pollard, M.; Thelen, J.J.; Paddock, T.; Salas, J.; Savage, L.; Milcamps, A.; Mhaske, V.B.; Cho, Y.; et al. Arabidopsis Genes Involved in Acyl Lipid Metabolism. A 2003 Census of the Candidates, a Study of the Distribution of Expressed Sequence Tags in Organs, and a Web-based database. Plant Physiol. 2003, 132, 681–697. [Google Scholar] [CrossRef]
- Duan, W.; Shi-Mei, Y.; Zhi-Wei, S.; Jing, X.; De-Gang, Z.; Hong-Bin, W.; Qi, S. Genome-Wide Analysis of the Fatty Acid Desaturase Gene Family Reveals the Key Role of PfFAD3 in α-Linolenic Acid Biosynthesis in Perilla Seeds. Front. Genet. 2021, 12, 735862. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C.; Shen, H.-B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van De Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.-B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Hinsinger, D.; Basak, D.; Gaudeul, J.; Cruaud, M.; Bertolino, C.; Bousquet, J. The phylogeny and biogeographic history of ashes (Fraxinus, Oleaceae) highlight the roles of migration and vicariance in the diversification of temperate trees. PLoS ONE 2013, 8, e80431. [Google Scholar] [CrossRef]
- Li, M.; Zhang, D.; Gao, Q.; Luo, Y.; Zhang, H.; Ma, B.; Chen, C.; Whibley, A.; Zhang, Y.; Cao, Y.; et al. Genome structure and evolution of Antirrhinum majus L. Nat. Plants 2019, 5, 174–183. [Google Scholar] [CrossRef]
- Melvin, P.; Prabhu, S.A.; Anup, C.P.; Shailasree, S.; Shetty, H.S.; Kini, K.R. Involvement of mitogen-activated protein kinase signalling in pearl millet-downy mildew interaction. Plant Sci. 2014, 214, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Jagodzik, P.; Tajdel-Zielinska, M.; Ciesla, A.; Marczak, M.; Ludwikow, A. Mitogen-Activated Protein Kinase Cascades in Plant Hormone Signaling. Front. Plant Sci. 2018, 9, 1387. [Google Scholar] [CrossRef] [PubMed]
- Bigeard, J.; Hirt, H. Nuclear Signaling of Plant MAPKs. Front. Plant Sci. 2018, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Oztas, O.; Selby, C.P.; Sancar, A.; Adebali, O. Genome-wide excision repair in Arabidopsis is coupled to transcription and reflects circadian gene expression patterns. Nat. Commun. 2018, 9, 1503. [Google Scholar] [CrossRef]
- Sharma, B.; Joshi, D.; Yadav, P.K.; Gupta, A.K.; Bhatt, T.K. Role of Ubiquitin-Mediated Degradation System in Plant Biology. Front. Plant Sci. 2016, 7, 806. [Google Scholar] [CrossRef] [PubMed]
- Kurepa, J.; Wang, S.; Li, Y.; Smalle, J. Proteasome regulation, plant growth and stress tolerance. Plant Signal. Behav. 2009, 4, 924–927. [Google Scholar] [CrossRef]
- Huang, R.; Liu, M.; Gong, G.; Wu, P.; Bai, M.; Qin, H.; Wang, G.; Liao, H.; Wang, X.; Li, Y.; et al. BLISTER promotes seed maturation and fatty acid biosynthesis by interacting with WRINKLED1 to regulate chromatin dynamics in Arabidopsis. Plant Cell 2022, 34, 2242–2265. [Google Scholar] [CrossRef]
- Sánchez, R.; González-Thuillier, I.; Venegas-Calerón, M.; Garcés, R.; Salas, J.J.; Martínez-Force, E. The Sunflower WRINKLED1 Transcription Factor Regulates Fatty Acid Biosynthesis Genes through an AW Box Binding Sequence with a Particular Base Bias. Plants 2022, 11, 972. [Google Scholar] [CrossRef]
- Wang, L.; Wang, C.; Liu, X.; Cheng, J.; Li, S.; Zhu, J.-K.; Gong, Z. Peroxisomal β-oxidation regulates histone acetylation and DNA methylation in Arabidopsis. Proc. Natl. Acad Sci. USA 2019, 116, 10576–10585. [Google Scholar] [CrossRef]
- Bates, P.D. The plant lipid metabolic network for assembly of diverse triacylglycerol molecular species. Adv. Bot. Res. 2022, 101, 225–252. [Google Scholar] [CrossRef]
- Yu, W.L.; Ansari, W.; Schoepp, N.G.; Hannon, M.J.; Mayfield, S.P.; Burkart, M.D. Modifications of the metabolic pathways of lipid and triacylglycerol production in microalgae. Microb. Cell Factories 2011, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.P.d.S.e.; da Costa, W.A.; Salazar, M.d.L.A.R.; Bezerra, P.D.N.; Pires, F.C.S.; Ferreira, M.C.R.; Menezes, E.G.O.; Urbina, G.R.O.; Barbosa, J.R.; Nunes, d.C.R. Commercial and therapeutic potential of plant-based fatty acids. In Biochemistry and Health Benefits of Fatty Acids; IntechOpen: London, UK, 2018; Volume 5, pp. 73–90. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, S.-H.; Lee, M.H.; Lee, J.-H.; Yu, Y.; Lee, J.; Kim, T.-H. The Genome of the Korean Island-Originated Perilla citriodora ‘Jeju17’ Sheds Light on Its Environmental Adaptation and Fatty Acid and Lipid Production Pathways. Genes 2023, 14, 1898. https://doi.org/10.3390/genes14101898
Bae S-H, Lee MH, Lee J-H, Yu Y, Lee J, Kim T-H. The Genome of the Korean Island-Originated Perilla citriodora ‘Jeju17’ Sheds Light on Its Environmental Adaptation and Fatty Acid and Lipid Production Pathways. Genes. 2023; 14(10):1898. https://doi.org/10.3390/genes14101898
Chicago/Turabian StyleBae, Seon-Hwa, Myoung Hee Lee, Jeong-Hee Lee, Yeisoo Yu, Jundae Lee, and Tae-Ho Kim. 2023. "The Genome of the Korean Island-Originated Perilla citriodora ‘Jeju17’ Sheds Light on Its Environmental Adaptation and Fatty Acid and Lipid Production Pathways" Genes 14, no. 10: 1898. https://doi.org/10.3390/genes14101898