

Association Analysis of Tiller-Related Traits with EST-SSR Markers in Psathyrostachys juncea


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Tiller Related Traits Phenotyping
2.2. EST-SSR Markers Development for P. juncea
2.3. Population Genotyping with EST-SSR Markers
2.4. Linkage Disequilibrium, Population Structure and Association Analysis
3. Results
3.1. Phenotypic Analysis of Tiller-Related Traits
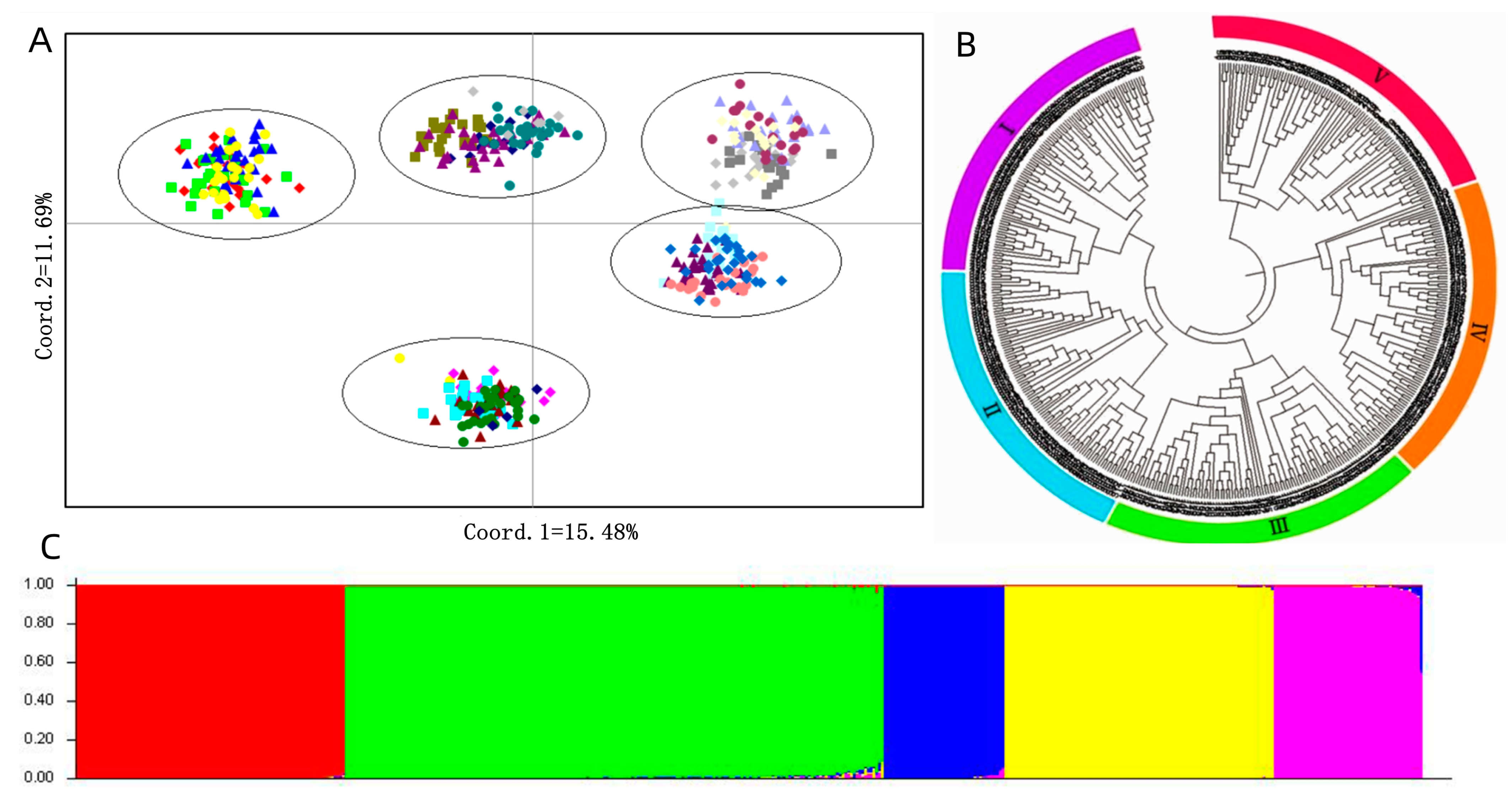
3.2. The Polymorphism of SSR Markers, Genetic Diversity and Population Structure Analysis
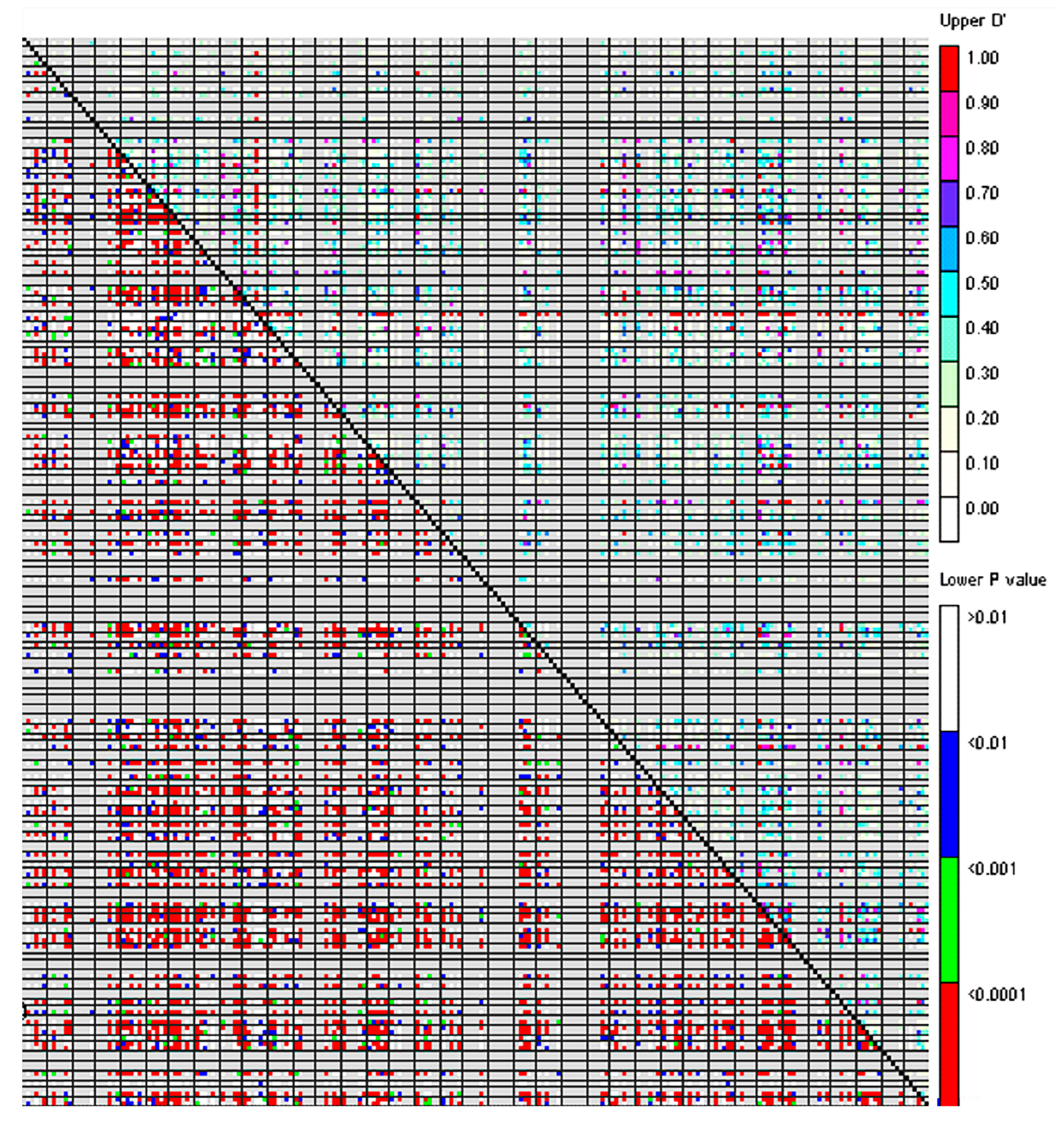
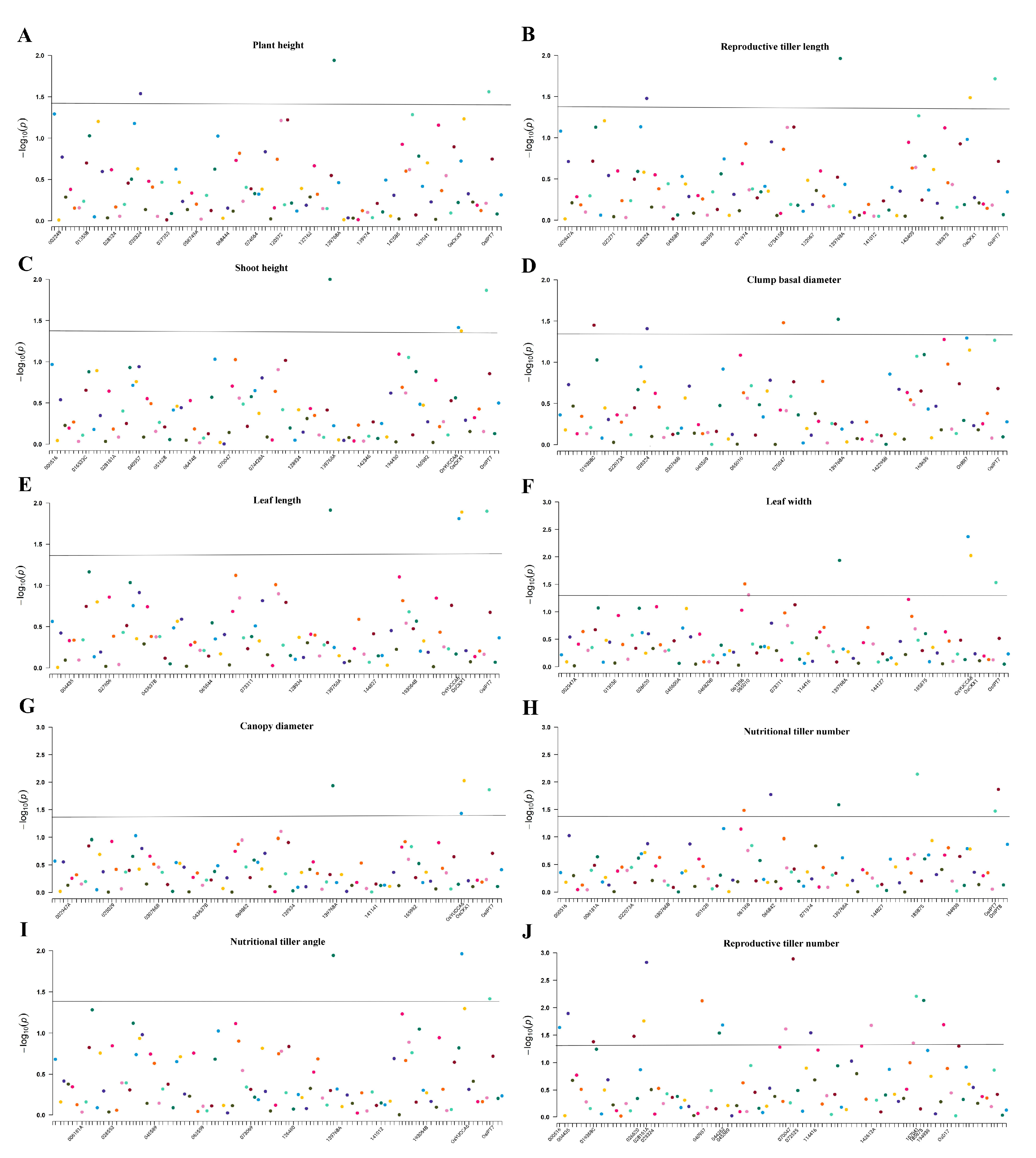
3.3. Linkage Disequilibrium and Association Analysis of the P. juncea Population
4. Discussion
4.1. Phenotyping Analysis of the P. juncea Population
4.2. Genetic Diversity Analysis of P. juncea Material
4.3. Population Structure of P. juncea Germplasm Resources
4.4. Linkage Disequilibrium Analysis of P. juncea Population
4.5. Association Analysis of P. juncea
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wei, J.Z.; Campbell, W.F.; Wang, R.R.C. Genetic Variability in Russian Wildrye (Psathyrostachys juncea) Assessed by RAPD. Genet. Resour. Crop Evol. 1997, 44, 117–125. [Google Scholar] [CrossRef]
- Wang, Q.Z.; Zhang, T.J.; Cui, J.; Wang, X.G.; Zhou, H.; Han, J.G.; Ren, E.; Gislum, R. Path and Ridge Regression Analysis of Seed Yield and Seed Yield Components of Russian Wildrye (Psathyrostachys juncea Nevski) under Field Conditions. PLoS ONE 2011, 6, e18245. [Google Scholar] [CrossRef]
- John, D.B.; Ronald, E.R. Development and Vigor of Diploid and Tetraploid Russian Wildrye Seedlings. J. Range Manag. 1997, 50, 80–84. [Google Scholar]
- Wang, Z.Y.; Bell, J.; Lehmann, D. Transgenic Russian Wildrye (Psathyrostachys juncea) Plants Obtained by Biolistic Transformation of Embryogenic Suspension Cells. Plant Cell Rep. 2004, 22, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Ratzlaff, C.D. Performance of Some Native and Introduced Grasses in a Semiarid Region of Western Canada. Can. J. Plant Sci. 1989, 69, 251–254. [Google Scholar] [CrossRef]
- Jefferson, P.; Muri, R. Competition, Light Quality and Seedling Growth of Russian Wildrye Grass (Psathyrostachys juncea). Acta Agron. Hung. 2007, 55, 49–60. [Google Scholar] [CrossRef]
- Tan, Q.Y.; Zhu, H.T.; Liu, H.; Ni, Y.R.; Wu, S.Z.; Luan, X.; Liu, J.W.; Yang, W.F.; Yang, Z.F.; Zeng, R.Z.; et al. Fine Mapping of QTLs for Stigma Exsertion Rate from Oryza glaberrima by Chromosome Segment Substitution. Rice Sci. 2022, 29, 55–66. [Google Scholar]
- Ding, Y.; Zhang, X.; Ma, Q.; Li, F.; Tao, R.; Zhu, M.; Li, C.; Zhu, X.; Guo, W.; Ding, J. Tiller Fertility Is Critical for Improving Grain Yield, Photosynthesis, and Nitrogen Efficiency in Wheat. J. Integr. Agric. 2023, 22, 2054–2066. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, R.; Hu, Y.; Chen, J. Genome-Wide Linkage Mapping of Quantitative Trait Loci for Late-Season Physiological and Agronomic Traits in Spring Wheat under Irrigated Conditions. Agronomy 2018, 8, 60. [Google Scholar] [CrossRef]
- Yin, Y.; An, W.; Zhao, J.; Li, Y.; Fan, Y.; Chen, J.; Cao, Y.; Zhan, X. Constructing the Wolfberry (Lycium spp.) Genetic Linkage Map Using AFLP and SSR Markers. J. Integr. Agric. 2022, 21, 131–138. [Google Scholar] [CrossRef]
- Alahmad, S.; El Hassouni, K.; Bassi, F.M.; Dinglasan, E.; Youssef, C.; Quarry, G.; Aksoy, A.; Mazzucotelli, E.; Juhász, A.; Able, J.A.; et al. A Major Root Architecture QTL Responding to Water Limitation in Durum Wheat. Front. Plant Sci. 2019, 10, 436. [Google Scholar] [CrossRef]
- Sukumaran, S.; Reynolds, M.P.; Sansaloni, C. Genome-Wide Association Analyses Identify QTL Hotspots for Yield and Component Traits in Durum Wheat Grown under Yield Potential, Drought, and Heat Stress Environments. Front. Plant Sci. 2018, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of Linkage Disequilibrium in Plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, I.; Desta, Z.A.; Tripathi, R.K.; Beattie, A.; Badea, A.; Singh, J. Interaction and association analysis of malting related traits in barley. PLoS ONE 2023, 17, e0283763. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lu, X.L.; Wang, C.J.; Shen, L.; Dai, L.P.; He, J.L.; Yang, L.; Li, P.Y.; Hong, Y.; Zhang, Q.; et al. Genome-wide association study and transcriptome analysis reveal new QTL and candidate genes for nitrogen-deficiency tolerance in rice. Crop J. 2022, 10, 942–951. [Google Scholar] [CrossRef]
- Hosseini, M.S.; Ebrahimi, M.; Samsampour, D.; Abadía, J.; Khanahmadi, M.; Amirian, R.; Ghafoori, I.N.; Ghaderi-Zefrehei, M.; Gogorcena, Y. Association Analysis and Molecular Tagging of Phytochemicals in the Endangered Medicinal Plant Licorice (Glycyrrhiza glabra L.). Phytochemistry 2021, 183, 112629. [Google Scholar] [CrossRef]
- Mehrabi, A.A.; Pour-Aboughadareh, A.; Mansouri, S.; Hosseini, A. Genome-Wide Association Analysis of Root System Architecture Features and Agronomic Traits in Durum Wheat. Mol. Breed. 2020, 40, 55. [Google Scholar] [CrossRef]
- Holland, J. Genetic Architecture of Complex Traits in Plants. Curr. Opin. Plant Biol. 2007, 10, 156–161. [Google Scholar] [CrossRef]
- He, F.; Wei, C.; Zhang, Y.; Long, R.; Li, M.; Wang, Z.; Yang, Q.; Kang, J.; Chen, L. Genome-Wide Association Analysis Coupled With Transcriptome Analysis Reveals Candidate Genes Related to Salt Stress in Alfalfa (Medicago sativa L.). Front. Plant Sci. 2022, 12, 826584. [Google Scholar] [CrossRef]
- Qi, T.T.; Liu, H.Y.; Si, E.J.; Meng, Y.X.; Wang, C.J.; Wang, H.J. SSR Markers and Linkage Unbalance Analysis of Different Barley Materials. Mol. Plant Breed. 2021, 9, 1–18. [Google Scholar]
- Li, C.; Wang, Y.; Ai, N.; Li, Y.; Song, J. A Genome-wide Association Study of Early-maturation Traits in Upland Cotton Based on the CottonSNP80K Array. J. Integr. Plant Biol. 2018, 60, 970–985. [Google Scholar] [CrossRef]
- Ogrodowicz, P.; Mikołajczak, K.; Kempa, M.; Mokrzycka, M.; Krajewski, P.; Kuczyńska, A. Genome-Wide Association Study of Agronomical and Root-Related Traits in Spring Barley Collection Grown under Field Conditions. Front. Plant Sci. 2023, 14, 1077631. [Google Scholar] [CrossRef] [PubMed]
- Daudi, H.; Shimelis, H.; Mathew, I.; Oteng-Frimpong, R.; Ojiewo, C.; Varshney, R.K. Genetic Diversity and Population Structure of Groundnut (Arachis hypogaea L.) Accessions Using Phenotypic Traits and SSR Markers: Implications for Rust Resistance Breeding. Genet. Resour. Crop Evol. 2021, 68, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Mishal, K.; Rehana, K.; Armghan, S.; Ghulam, M.A.; Sania, B. Screening and validation of salt-stress responsive cg-SSR markers in wheat (Triticum aestivum L.) germplasm of Pakistan. Mol. Biol. Rep. 2023, 50, 5931–5940. [Google Scholar]
- Sainan, M.; Han, C.Y.; Jie Zhou, J.; Hu, R.C.; Jiang, X.; Wu, F.F.; Tian, K.; Nie, G.; Zhang, X.Q. Fingerprint identification of white clover cultivars based on SSR molecular markers. Mol. Biol. Rep. 2020, 47, 8513–8521. [Google Scholar]
- Li, F.; Peng, J. Genetic and Association Mapping Study of English Grain Aphid Resistance and Tolerance in Bread Wheat Germplasm. J. Integr. Agric. 2014, 13, 40–53. [Google Scholar] [CrossRef]
- Pour-Aboughadareh, A.; Jadidi, O.; Shooshtari, L.; Poczai, P.; Mehrabi, A.A. Association Analysis for Some Biochemical Traits in Wild Relatives of Wheat under Drought Stress Conditions. Genes 2022, 13, 1491. [Google Scholar] [CrossRef]
- Breseghello, F.; Sorrells, M.E. Association Mapping of Kernel Size and Milling Quality in Wheat (Triticum aestivum L.) Cultivars. Genetics 2006, 172, 1165–1177. [Google Scholar] [CrossRef]
- Yu, X.; Bai, G.; Liu, S.; Luo, N.; Wang, Y.; Richmond, D.S.; Pijut, P.M.; Jackson, S.A.; Yu, J.; Jiang, Y. Association of Candidate Genes with Drought Tolerance Traits in Diverse Perennial Ryegrass Accessions. J. Exp. Bot. 2013, 64, 1537–1551. [Google Scholar] [CrossRef]
- Yu, X.; Bai, G.; Luo, N.; Chen, Z.; Liu, S.; Liu, J.; Warnke, S.E.; Jiang, Y. Association of Simple Sequence Repeat (SSR) Markers with Submergence Tolerance in Diverse Populations of Perennial Ryegrass. Plant Sci. 2011, 180, 391–398. [Google Scholar] [CrossRef]
- Nie, G.; Tang, L.; Zhang, Y.J.; Huang, L.K.; Ma, X.; Cao, X.; Pan, L.; Zhang, X.; Zhang, X.Q. Development of SSR Markers Based on Transcriptome Sequencing and Association Analysis with Drought Tolerance in Perennial Grass Miscanthus from China. Front. Plant Sci. 2017, 8, 801. [Google Scholar] [CrossRef]
- Yan, H.D.; Zhang, Y.; Zeng, B.; Yin, G.H.; Zhang, X.Q.; Ji, Y.; Huang, L.K.; Jiang, X.M.; Liu, X.C.; Peng, Y. Genetic Diversity and Association of EST-SSR and SCoT Markers with Rust Traits in Orchardgrass (Dactylis glomerata L.). Molecules 2016, 21, 66. [Google Scholar] [CrossRef] [PubMed]
- Ming, S.; Zhang, C.L.; Zhang, X.Q.; Fan, Y.; Fu, K.X.; Wu, W.D.; Bai, S.Q.; Zhang, J.B.; Peng, Y.; Huang, L.K. AFLP assessment of genetic variability and relationships in an Asian wild germplasm collection of Dactylis glomerata L. Comptes Rendus Biol. 2017, 340, 145–155. [Google Scholar]
- Li, Z.; Yun, L.; Gao, Z.; Wang, T.; Ren, X.; Zhao, Y. EST-SSR Primer Development and Genetic Structure Analysis of Psathyrostachys juncea Nevski. Front. Plant Sci. 2022, 13, 837787. [Google Scholar] [CrossRef]
- Khan, M.A.; Von Witzke-Ehbrecht, S.; Maass, B.L.; Becker, H.C. Relationships among Different Geographical Groups, Agro-Morphology, Fatty Acid Composition and RAPD Marker Diversity in Safflower (Carthamus tinctorius). Genet. Resour. Crop Evol. 2009, 56, 19–30. [Google Scholar] [CrossRef]
- Gao, Z.; Yun, L.; Li, Z.; Liu, Q.; Zhang, C.; Ma, Y.; Shi, F. Hybrid Purity Identification Using EST-SSR Markers and Heterosis Analysis of Quantitative Traits of Russian Wildrye. Peer J. 2022, 10, e14442. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Soft. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software Structure: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Annicchiarico, P. Alfalfa Forage Yield and Leaf/Stem Ratio: Narrow-Sense Heritability, Genetic Correlation, and Parent Selection Procedures. Euphytica 2015, 205, 409–420. [Google Scholar] [CrossRef]
- Gao, Z.; Wang, Y.; Tian, G.; Zhao, Y.; Li, C.; Cao, Q.; Han, R.; Shi, Z.; He, M. Plant Height and Its Relationship with Yield in Wheat under Different Irrigation Regime. Irrig. Sci. 2020, 38, 365–371. [Google Scholar] [CrossRef]
- Ma, L.; Qing, C.; Zhang, M.; Zou, C.; Pan, G.; Shen, Y. GWAS with a PCA Uncovers Candidate Genes for Accumulations of Microelements in Maize Seedlings. Physiol. Plant. 2021, 172, 2170–2180. [Google Scholar] [CrossRef]
- Li, X.H.; Wang, K.J. Research Progress of Genetic Diversity in Wild Soybean (Glycine soja Siebold & Zucc.). J. Plant Genet. Resour. 2020, 21, 1344–1356. [Google Scholar]
- Ellegren, H.; Galtier, N. Determinants of Genetic Diversity. Nat. Rev. Genet. 2016, 17, 422–433. [Google Scholar] [CrossRef]
- Yang, F.; Wan, H.; Li, J.; Wang, Q.; Yang, N.; Zhu, X.; Liu, Z.; Yang, Y.; Ma, W.; Fan, X.; et al. Pentaploidization Enriches the Genetic Diversity of Wheat by Enhancing the Recombination of AB Genomes. Front. Plant Sci. 2022, 13, 883868. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Danting, S.; Wang, J.; Sadiq, H.; Rasheed, A.; He, Z.; Li, H. Genetic Diversity and Selection Signatures in Synthetic-Derived Wheats and Modern Spring Wheat. Front. Plant Sci. 2022, 13, 877496. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Feuerstein, U.; Luesink, W.; Schulze, S.; Asp, T.; Studer, B.; Becker, H.C.; Dehmer, K.J. DArT, SNP, and SSR Analyses of Genetic Diversity in Lolium Perenne L. Using Bulk Sampling. BMC Genet. 2018, 19, 10. [Google Scholar] [CrossRef]
- Verwimp, C.; Ruttink, T.; Muylle, H.; Van Glabeke, S.; Cnops, G.; Quataert, P.; Honnay, O.; Roldán-Ruiz, I. Temporal Changes in Genetic Diversity and Forage Yield of Perennial Ryegrass in Monoculture and in Combination with Red Clover in Swards. PLoS ONE 2018, 13, e0206571. [Google Scholar] [CrossRef]
- Sayed, M.R.I.; Alshallash, K.S.; Safhi, F.A.; Alatawi, A.; ALshamrani, S.M.; Dessoky, E.S.; Althobaiti, A.T.; Althaqafi, M.M.; Gharib, H.S.; Shafie, W.W.M.; et al. Genetic Diversity, Analysis of Some Agro-Morphological and Quality Traits and Utilization of Plant Resources of Alfalfa. Genes 2022, 13, 1521. [Google Scholar] [CrossRef] [PubMed]
- Pour-Aboughadareh, A.; Mahmoudi, M.; Moghaddam, M.; Ahmadi, J.; Mehrabi, A.A.; Alavikia, S.S. Agro-Morphological and Molecular Variability in Triticum Boeoticum Accessions from Zagros Mountains, Iran. Genet. Resour. Crop Evol. 2017, 64, 545–556. [Google Scholar] [CrossRef]
- Nie, G.; Huang, T.; Ma, X.; Huang, L.; Peng, Y.; Yan, Y.; Li, Z.; Wang, X.; Zhang, X. Genetic Variability Evaluation and Cultivar Identification of Tetraploid Annual Ryegrass Using SSR Markers. Peer J. 2019, 7, e7742. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, H.X.; Li, Y.S.; Huang, Z.H.; Yin, Z.J.; Yang, C.X.; Du, Z.Q. GWLD: An R package for genome-wide linkage disequilibrium analysis. G3 Genes Genomes Genet. 2023, 13, jkad154. [Google Scholar] [CrossRef]
- Pablo, F.R.; Adelina, O.L.; Ana, L.A.; Carolina, S.P.; Cristian, A.G.; Susanne, D.; Viviana, E. Linkage disequilibrium patterns, population structure and diversity analysis in a worldwide durum wheat collection including Argentinian genotypes. BMC Genom. 2021, 22, 233. [Google Scholar]
- Wang, H.; Zhu, S.; Dang, X.; Liu, E.; Hu, X.; Eltahawy, M.S.; Zaid, I.U.; Hong, D. Favorable Alleles Mining for Gelatinization Temperature, Gel Consistency and Amylose Content in Oryza sativa by Association Mapping. BMC Genet. 2019, 20, 34. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Gou, Y.; Heng, Y.; Ding, W.; Li, Y.; Zhou, D.; Li, X.; Liang, C.; Wu, C.; Wang, H.; et al. Targeted Manipulation of Grain Shape Genes Effectively Improves Outcrossing Rate and Hybrid Seed Production in Rice. Plant Biotechnol. J. 2023, 21, 381–390. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for Association Mapping of Complex Traits in Diverse Samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; McCarty, J.C.; Jenkins, J.N.; Saha, S. QTLs for Node of First Fruiting Branch in a Cross of an Upland Cotton, Gossypium Hirsutum L., Cultivar with Primitive Accession Texas 701. Euphytica 2008, 163, 113–122. [Google Scholar] [CrossRef]
- He, G.; Luo, X.; Tian, F.; Li, K.; Zhu, Z.; Su, W.; Qian, X.; Fu, Y.; Wang, X.; Sun, C.; et al. Haplotype Variation in Structure and Expression of a Gene Cluster Associated with a Quantitative Trait Locus for Improved Yield in Rice. Genome Res. 2006, 16, 618–626. [Google Scholar] [CrossRef]
- Liu, J.; Wang, J.; Wang, H.; Wang, W.; Zhou, R.; Mei, D.; Cheng, H.; Yang, J.; Raman, H.; Hu, Q. Multigenic Control of Pod Shattering Resistance in Chinese Rapeseed Germplasm Revealed by Genome-Wide Association and Linkage Analyses. Front. Plant Sci. 2016, 7, 1058. [Google Scholar] [CrossRef]
- Si, F.F.; Fan, F.F.; Wei, X.; He, S.H.; Li, X.L.; Peng, X.J.; Li, S.Q. Quantitative Trait Locus Mapping of High Photosynthetic Efficiency and Biomass in Oryza longistaminata. Rice Sci. 2022, 29, 569–578. [Google Scholar]
- Wei, W.; Mesquita, A.C.O.; Figueiró, A.D.A.; Wu, X.; Manjunatha, S.; Wickland, D.P.; Hudson, M.E.; Juliatti, F.C.; Clough, S.J. Genome-Wide Association Mapping of Resistance to a Brazilian Isolate of Sclerotinia sclerotiorum in Soybean Genotypes Mostly from Brazil. BMC Genom. 2017, 18, 849. [Google Scholar] [CrossRef] [PubMed]
- Elakhdar, A.; EL-Sattar, M.A.; Amer, K.; Rady, A.; Kumamaru, T. Population Structure and Marker–Trait Association of Salt Tolerance in Barley (Hordeum vulgare L.). Comptes Rendus Biol. 2016, 339, 454–461. [Google Scholar] [CrossRef]
- Ghorbanipour, A.; Rabiei, B.; Rahmanpour, S.; Khodaparast, S.A. Association Analysis of Charcoal Rot Disease Resistance in Soybean. Plant Pathol. J. 2019, 35, 189–199. [Google Scholar] [CrossRef]
- Arabi, M.I.E.; Daoude, A.A.; Mokrani, L.; Shoaib, A.; Jawhar, M. Identification of AFLP Markers Associated with Spot Blotch Resistance through Single Marker Analysis in Barley (Hordeum vulgare L.). Cereal Res. Commun. 2021, 49, 285–290. [Google Scholar] [CrossRef]
- Rani, R.; Raza, G.; Ashfaq, H.; Rizwan, M.; Shimelis, H.; Tung, M.H.; Arif, M. Analysis of Genotype × Environment Interactions for Agronomic Traits of Soybean (Glycine max [L.] Merr.) Using Association Mapping. Front. Genet. 2023, 13, 1090994. [Google Scholar] [CrossRef]
- Song, J.; Pei, W.; Wang, N.; Ma, J.; Xin, Y.; Yang, S.; Wang, W.; Chen, Q.; Zhang, J.; Yu, J.; et al. Transcriptome Analysis and Identification of Genes Associated with Oil Accumulation in Upland Cotton. Physiol. Plant. 2022, 174, e13701. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Jia, J.L.; Wang, J.M.; Ren, L.; Lv, Z.Y.; Zhu, H.Q.; Ma, H.; Yang, L.N.; Li, Z.R. Analysis of Genetic Diversity and Association with Agronomic Traits in Barley(Hordeum vulgare L.)Introduced from Abroad Using SSR Markers. J. Triticeae Crops 2017, 37, 197–204. [Google Scholar]
- Ober, E.S.; Howell, P.; Thomelin, P.; Kouidri, A. The Importance of Accurate Developmental Staging. J. Exp. Bot. 2020, 71, 3375–3379. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, S.; Liang, Y.; Zhang, R.; Liu, L.; Qin, P.; Zhang, Z.; Wang, Y.; Zhou, J.; Tang, X.; et al. Loss-function Mutants of OsCKX Gene Family Based on CRISPR-Cas Systems Revealed Their Diversified Roles in Rice. Plant Genome 2023, 16, e20283. [Google Scholar] [CrossRef]
- Ghosh, A.; Shah, M.N.A.; Jui, Z.S.; Saha, S.; Fariha, K.A.; Islam, T. Evolutionary Variation and Expression Profiling of Isopentenyl Transferase Gene Family in Arabidopsis thaliana L. and Oryza sativa L. Plant Gene 2018, 15, 15–27. [Google Scholar] [CrossRef]
- Prakash, S.; Rai, R.; Zamzam, M.; Ahmad, O.; Peesapati, R.; Vijayraghavan, U. OsbZIP47 Is an Integrator for Meristem Regulators During Rice Plant Growth and Development. Front. Plant Sci. 2022, 13, 865928. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, J.; Ding, Y.; Wang, Q.; Li, G.; Wang, S. Auxin Inhibits the Outgrowth of Tiller Buds in Rice (Oryza sativa L.) by Downregulating OsIPT Expression and Cytokinin Biosynthesis in Nodes. Aust. J. Crop Sci. 2011, 5, 169–174. [Google Scholar]
- Liu, Y.; Ding, Y.; Wang, Q.; Meng, D.; Wang, S. Effects of Nitrogen and 6-Benzylaminopurine on Rice Tiller Bud Growth and Changes in Endogenous Hormones and Nitrogen. Crop Sci. 2011, 51, 786–792. [Google Scholar] [CrossRef]
- Li, Z.; Yun, L.; Ren, X.; Shi, F.; Mi, F. Analysis of Controlling Genes for Tiller Growth of Psathyrostachys Juncea Based on Transcriptome Sequencing Technology. BMC Plant Biol. 2022, 22, 456. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.M.; Yun, L.; Ai, Q.; Li, Z.; Zhao, Q.; Shi, F.L. Cloning and expression analysis of YUCCA gene in Psathyrostachys juncea. J. Northwest A F Univ. Nat. Sci. Ed. 2023, accepted. [Google Scholar]
- Ren, X.M.; Yun, L.; Ai, Q.; Li, Z.; Zhao, Q.; Shi, F.L. Subcellular Localization, Overexpression Vector Construction and Identification of IPT Gene in Psathyrostachys juncea. Acta Bot. Boreali-Occident. Sin. 2023. accepted. [Google Scholar]
- Ren, X.M.; Yun, L.; Li, Z.; Ai, Q.; Zhao, Q.; Shi, F.L. Cloning and Expression Analysis of Cytokinin Regulatory Gene CKX4 in Psathyrostachys juncea. Acta Agrestia Sin. 2023, 9, 1–16. [Google Scholar]
- Jia, Z.Y. Creation of New Germplasm and Association Analysis of Main Agronomic Traits in Elymus sibiricus L.; Inner Mongolia Agricultural University: Hohhot, China, 2021. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Environment | Range | Mean | SD | Coefficient of Variation (%) |
|---|---|---|---|---|---|
| Plant height | Hohhot | 19.00~91.73 | 60.91 | 14.82 | 24.33 |
| Baotou | 16.00~99.67 | 70.51 | 12.33 | 17.49 | |
| BLUE | 14.44~95.13 | 65.21 | 11.74 | 18.01 | |
| Reproductive tiller length | Hohhot | 23.17~107.43 | 72.19 | 14.77 | 20.46 |
| Baotou | 25.00~111.08 | 82.90 | 13.56 | 16.36 | |
| BLUE | 22.16~100.77 | 76.92 | 12.61 | 16.39 | |
| Shoot height | Hohhot | 12.60~60.90 | 30.88 | 8.07 | 26.15 |
| Baotou | 12.75~61.17 | 39.32 | 6.69 | 17.03 | |
| BLUE | 12.52~53.10 | 34.84 | 5.50 | 15.79 | |
| Clump basal diameter | Hohhot | 15.53~88.13 | 55.16 | 11.56 | 20.96 |
| Baotou | 15.50~108.67 | 66.38 | 17.65 | 26.59 | |
| BLUE | 11.09~97.06 | 60.23 | 12.80 | 21.25 | |
| Leaf length | Hohhot | 15.56~50.05 | 33.56 | 5.50 | 16.39 |
| Baotou | 20.33~52.73 | 40.46 | 5.55 | 13.72 | |
| BLUE | 20.08~47.47 | 36.86 | 4.59 | 12.45 | |
| Leaf width | Hohhot | 0.24~0.60 | 0.42 | 0.06 | 13.98 |
| Baotou | 0.23~0.60 | 0.41 | 0.05 | 12.93 | |
| BLUE | 0.26~0.56 | 0.42 | 0.05 | 11.04 | |
| Canopy diameter | Hohhot | 25.53~102.75 | 66.79 | 12.55 | 18.78 |
| Baotou | 31.50~105.83 | 74.29 | 12.36 | 16.64 | |
| BLUE | 33.34~96.02 | 70.12 | 10.44 | 14.89 | |
| Nutritional tiller number | Hohhot | 17.17~453.67 | 152.36 | 61.49 | 40.36 |
| Baotou | 18.00~426.00 | 202.19 | 69.92 | 34.58 | |
| BLUE | 15.36~397.67 | 174.90 | 53.51 | 30.60 | |
| Nutritional tiller angle | Hohhot | 27.50~83.17 | 56.44 | 9.16 | 16.23 |
| Baotou | 39.17~80.00 | 61.03 | 5.64 | 9.25 | |
| BLUE | 41.07~79.16 | 58.73 | 4.75 | 8.09 | |
| Reproductive tiller number | Hohhot | 4.00~182.00 | 45.46 | 31.20 | 68.64 |
| Baotou | 8.00~325.00 | 89.25 | 45.23 | 50.68 | |
| BLUE | 3.10~186.18 | 59.90 | 32.31 | 53.95 |
| Traits | Indicator Eigenvector | |||
|---|---|---|---|---|
| I | II | III | IV | |
| Plant height | 0.40 * | −0.16 | −0.10 | −0.29 |
| Reproductive tiller length | 0.36 * | −0.06 | −0.07 | −0.16 |
| Shoot height | 0.37 * | −0.27 | 0.28 | 0.34 |
| Clump basal diameter | 0.31 * | 0.07 | −0.21 | −0.01 |
| Leaf length | 0.28 | 0.30 * | 0.16 | 0.01 |
| Leaf width | 0.13 | 0.72 * | −0.07 | −0.06 |
| Canopy diameter | 0.30 | 0.23 * | 0.02 | −0.05 |
| Nutritional tiller number | 0.25 | −0.09 | 0.34 | 0.79 * |
| Nutritional tiller angle | 0.12 | −0.41 | 0.75 * | −0.24 |
| Reproductive tiller number | 0.27 | −0.22 | 0.40 * | −0.29 |
| Contribution (%) | 46.82 | 11.10 | 9.87 | 7.02 |
| Cumulative Contribution (%) | 46.82 | 57.92 | 67.79 | 74.81 |
| Sources of Variation | Degrees of Freedom | Sum of Squares | Mean Square | Variance of Components | Percentage Variation (%) | p Value | Fst |
|---|---|---|---|---|---|---|---|
| Among Pops | 20 | 18,235.035 | 911.752 | 18.957 | 34 | <0.001 | 0.341 |
| Among Individuals | 459 | 21,331.484 | 46.474 | 9.826 | 18 | <0.001 | |
| Within Individuals | 480 | 12,874.500 | 26.822 | 26.822 | 48 | <0.001 | |
| Total | 959 | 52,441.020 | 55.605 | 100 | <0.001 |
| Range of D′ Value | No. of Loci Pairs with Significant LD | Percentage of Loci Pairs with Significant LD (%) |
|---|---|---|
| 0~0.2 | 2292 | 39.02 |
| 0.2~0.4 | 2072 | 35.27 |
| 0.4~0.6 | 886 | 15.08 |
| 0.6~0.8 | 342 | 5.82 |
| 0.8~1 | 282 | 4.80 |
| Traits | Markers | p Value | R2/% | Traits | Markers | p Value | R2/% |
|---|---|---|---|---|---|---|---|
| Plant height | 028324 | 0.029 | 2.290 | Nutritional tiller number | 061356 | 0.033 | 2.230 |
| 139768A | 0.012 | 1.910 | 066842 | 0.017 | 2.530 | ||
| OsIPT7 | 0.028 | 7.840 | 139768A | 0.026 | 1.520 | ||
| Reproductive tiller length | 028324 | 0.033 | 2.190 | 185875 | 0.007 | 2.500 | |
| 139768A | 0.011 | 1.930 | OsIPT7 | 0.034 | 7.400 | ||
| OsCKX1 | 0.033 | 1.490 | OsIPT8 | 0.014 | 1.960 | ||
| OsIPT7 | 0.019 | 8.500 | Reproductive tiller number | 000516 | 0.023 | 1.060 | |
| Shoot height | 139768A | 0.010 | 1.940 | 004435 | 0.013 | 1.880 | |
| OsYUCCA6 | 0.039 | 1.430 | 019368C | 0.042 | 1.580 | ||
| OsCKX1 | 0.042 | 1.360 | 026620 | 0.033 | 1.950 | ||
| OsIPT7 | 0.014 | 9.150 | 028161A | 0.018 | 1.960 | ||
| Clump basal diameter | 019368C | 0.036 | 1.680 | 028324 | 0.002 | 4.130 | |
| 028324 | 0.039 | 2.060 | 040957 | 0.008 | 4.190 | ||
| 070047 | 0.033 | 3.200 | 044262 | 0.029 | 3.350 | ||
| 139768A | 0.030 | 1.500 | 045589 | 0.021 | 1.100 | ||
| Leaf length | 139768A | 0.012 | 1.870 | 070047 | 0.025 | 1.630 | |
| OsYUCCA6 | 0.016 | 1.840 | 072525 | 0.001 | 2.100 | ||
| OsCKX1 | 0.013 | 1.880 | 114416 | 0.029 | 1.550 | ||
| OsIPT7 | 0.013 | 9.210 | 142612A | 0.021 | 1.600 | ||
| Leaf width | 061356 | 0.031 | 2.390 | 167041 | 0.044 | 2.550 | |
| 065010 | 0.049 | 2.530 | 185875 | 0.006 | 2.670 | ||
| 139768A | 0.012 | 1.910 | 194938 | 0.007 | 2.620 | ||
| OsYUCCA6 | 0.004 | 2.420 | OsD17 | 0.020 | 1.840 | ||
| OsCKX1 | 0.010 | 2.030 | Canopy diameter | 139768A | 0.012 | 1.910 | |
| OsIPT7 | 0.029 | 7.650 | OsYUCCA6 | 0.037 | 1.470 | ||
| Nutritional tiller angle | 139768A | 0.011 | 1.890 | OsCKX1 | 0.009 | 2.030 | |
| OsYUCCA6 | 0.011 | 1.990 | OsIPT7 | 0.014 | 9.100 | ||
| OsIPT7 | 0.038 | 7.070 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Wang, T.; Yun, L.; Ren, X.; Wang, Y.; Shi, F. Association Analysis of Tiller-Related Traits with EST-SSR Markers in Psathyrostachys juncea. Genes 2023, 14, 1970. https://doi.org/10.3390/genes14101970
Li Z, Wang T, Yun L, Ren X, Wang Y, Shi F. Association Analysis of Tiller-Related Traits with EST-SSR Markers in Psathyrostachys juncea. Genes. 2023; 14(10):1970. https://doi.org/10.3390/genes14101970
Chicago/Turabian StyleLi, Zhen, Tian Wang, Lan Yun, Xiaomin Ren, Yong Wang, and Fengling Shi. 2023. "Association Analysis of Tiller-Related Traits with EST-SSR Markers in Psathyrostachys juncea" Genes 14, no. 10: 1970. https://doi.org/10.3390/genes14101970
APA StyleLi, Z., Wang, T., Yun, L., Ren, X., Wang, Y., & Shi, F. (2023). Association Analysis of Tiller-Related Traits with EST-SSR Markers in Psathyrostachys juncea. Genes, 14(10), 1970. https://doi.org/10.3390/genes14101970