
Novel Loss of Function Variants in CENPF Including a Large Intragenic Deletion in Patients with Strømme Syndrome

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Whole Exome Sequencing (WES) and Variant Verification
2.2. RNA Sequencing (RNA-seq) and Data Analysis
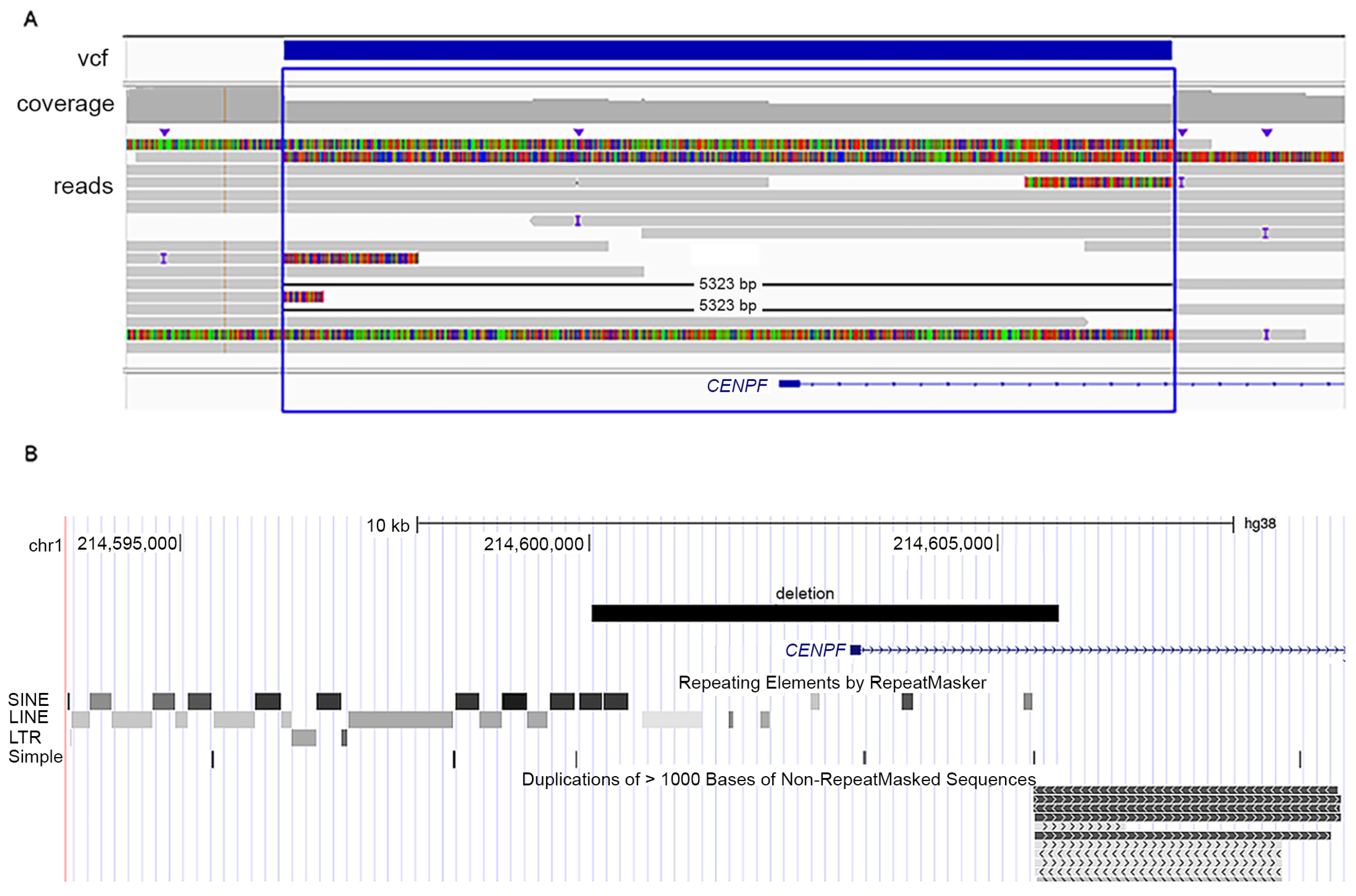
2.3. Long-Read Whole Genome Sequencing
3. Results
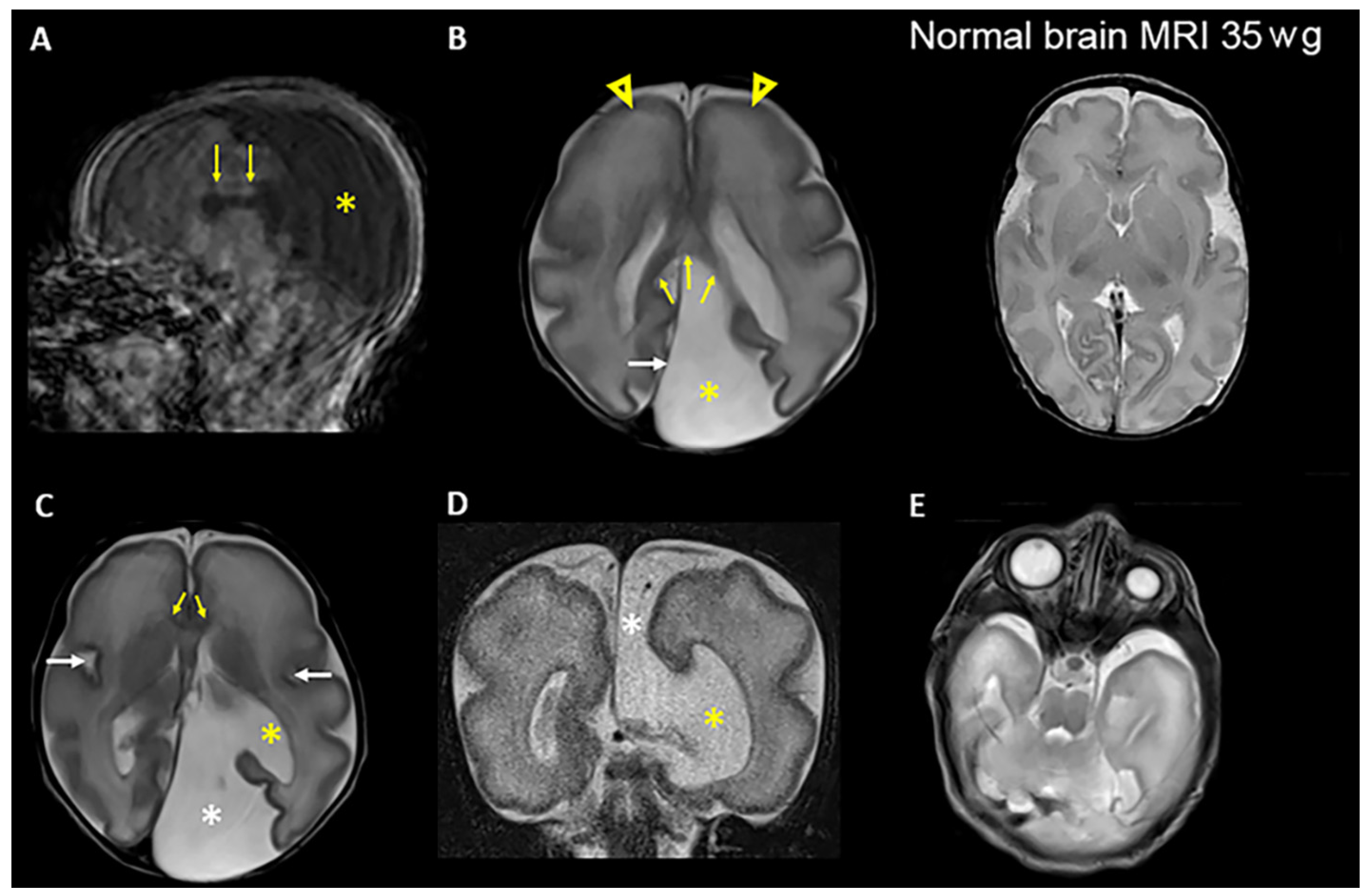
3.1. Clinical Presentations
3.2. Genetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strømme, P.; Dahl, E.; Flage, T.; Stene-Johansen, H. Apple peel intestinal atresia in siblings with ocular anomalies and microcephaly. Clin. Genet. 1993, 44, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Strømme, P.; Andersen, W. Developmental aspects in apple peel intestinal atresia-ocular anomalies-microcephaly syndrome. Clin. Genet. 1997, 52, 133. [Google Scholar] [CrossRef] [PubMed]
- Al-Dewik, N.; Mohd, H.; Al-Mureikhi, M.; Ali, R.; Al-Mesaifri, F.; Mahmoud, L.; Shahbeck, N.; El-Akouri, K.; Almulla, M.; Al Sulaiman, R.; et al. Clinical exome sequencing in 509 Middle Eastern families with suspected Mendelian diseases: The Qatari experience. Am. J. Med. Genet. Part A 2019, 179, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamed, M.H.; Kurdi, W.; Khan, R.; Tulbah, M.; AlNemer, M.; AlSahan, N.; AlMugbel, M.; Rafiullah, R.; Assoum, M.; Monies, D.; et al. Prenatal exome sequencing and chromosomal microarray analysis in fetal structural anomalies in a highly consanguineous population reveals a propensity of ciliopathy genes causing multisystem phenotypes. Hum. Genet. 2022, 141, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, M.; Alkhamis, W.H.; Bashiri, F.A.; Jamjoom, D.; Al-Nafisah, G.; Tahir, A.; Abdouelhoda, M. Expanding the phenotype and the genotype of Stromme syndrome: A novel variant of the CENPF gene and literature review. Eur. J. Med. Genet. 2020, 63, 103844. [Google Scholar] [CrossRef] [PubMed]
- Blue, G.M.; Mekel, M.; Das, D.; Troup, M.; Rath, E.; Ip, E.; Gudkov, M.; Perumal, G.; Harvey, R.P.; Sholler, G.F.; et al. Whole genome sequencing in transposition of the great arteries and associations with clinically relevant heart, brain and laterality genes. Am. Heart J. 2022, 244, 1–13. [Google Scholar] [CrossRef]
- Cappuccio, G.; Brillante, S.; Tammaro, R.; Pinelli, M.; De Bernardi, M.L.; Gensini, M.G.; Bijlsma, E.K.; Koopmann, T.T.; Hoffer, M.J.V.; McDonald, K.; et al. Biallelic variants in CENPF causing a phenotype distinct from Strømme syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 102–108. [Google Scholar] [CrossRef]
- Caridi, G.; Lugani, F.; Lerone, M.; Divizia, M.T.; Ghiggeri, G.M.; Verrina, E. Renal involvement and Strømme syndrome. Clin. Kidney J. 2021, 14, 439–441. [Google Scholar] [CrossRef]
- Filges, I.; Bruder, E.; Brandal, K.; Meier, S.; Undlien, D.E.; Waage, T.R.; Hoesli, I.; Schubach, M.; de Beer, T.; Sheng, Y.; et al. Strømme Syndrome Is a Ciliary Disorder Caused by Mutations in CENPF. Hum. Mutat. 2016, 37, 711. [Google Scholar] [CrossRef]
- Ho, S.; Luk, H.; Lo, I.F.M. The first case report of Strømme syndrome in a Chinese patient: Expanding the phenotype and literature review. Am. J. Med. Genet. Part A 2022, 188, 1626–1629. [Google Scholar] [CrossRef]
- Ho, S.K.L.; Leung, L.T.; Luk, H.M. Stromme Syndrome. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Maddirevula, S.; Alzahrani, F.; Al-Owain, M.; Al Muhaizea, M.A.; Kayyali, H.R.; AlHashem, A.; Rahbeeni, Z.; Al-Otaibi, M.; Alzaidan, H.I.; Balobaid, A.; et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med. 2019, 21, 736–742. [Google Scholar] [CrossRef]
- Ozkinay, F.; Atik, T.; Isik, E.; Gormez, Z.; Sagiroglu, M.; Sahin, O.A.; Corduk, N.; Onay, H. A further family of Stromme syndrome carrying CENPF mutation. Am. J. Med. Genet. Part A 2017, 173, 1668–1672. [Google Scholar] [CrossRef]
- Palombo, F.; Graziano, C.; Al Wardy, N.; Nouri, N.; Marconi, C.; Magini, P.; Severi, G.; La Morgia, C.; Cantalupo, G.; Cordelli, D.M.; et al. Autozygosity-driven genetic diagnosis in consanguineous families from Italy and the Greater Middle East. Hum. Genet. 2020, 139, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Asfahani, R.; Carroll, P.; Bicknell, L.; Lescai, F.; Bright, A.; Chanudet, E.; Brooks, A.; Christou-Savina, S.; Osman, G.; et al. The kinetochore protein, CENPF, is mutated in human ciliopathy and microcephaly phenotypes. J. Med. Genet. 2015, 52, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Winkfein, R.J.; Mack, G.; Rattner, J.B.; Yen, T.J. CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J. Cell Biol. 1995, 130, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Berto, A.; Doye, V. Regulation of Cenp-F localization to nuclear pores and kinetochores. Cell Cycle 2018, 17, 2122–2133. [Google Scholar] [CrossRef]
- Maton, G.; Edwards, F.; Lacroix, B.; Stefanutti, M.; Laband, K.; Lieury, T.; Kim, T.; Espeut, J.; Canman, J.C.; Dumont, J. Kinetochore components are required for central spindle assembly. Nat. Cell Biol. 2015, 17, 697–705. [Google Scholar] [CrossRef]
- Bellini, C.; Mazzella, M.; Arioni, C.; Fondelli, M.P.; Serra, G. “Apple-peel” intestinal atresia, ocular anomalies, and microcephaly syndrome: Brain magnetic resonance imaging study. Am. J. Med. Genet. 2002, 110, 176–178. [Google Scholar] [CrossRef]
- Epting, D.; Senaratne, L.D.S.; Ott, E.; Holmgren, A.; Sumathipala, D.; Larsen, S.M.; Wallmeier, J.; Bracht, D.; Frikstad, K.M.; Crowley, S.; et al. Loss of CBY1 results in a ciliopathy characterized by features of Joubert syndrome. Hum. Mutat. 2020, 41, 2179–2194. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.-Y.; Dillies, M.-A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Castori, M.; Laino, L.; Briganti, V.; Pedace, L.; Zampini, A.; Marconi, M.; Grammatico, B.; Buffone, E.; Grammatico, P. Jejunal atresia and anterior chamber anomalies: Further delineation of the Strømme syndrome. Eur. J. Med. Genet. 2010, 53, 149–152. [Google Scholar] [CrossRef]
- Keegan, C.E.; Vilain, E.; Mohammed, M.; Lehoczky, J.; Dobyns, W.B.; Archer, S.M.; Innis, J.W. Microcephaly, jejunal atresia, aberrant right bronchus, ocular anomalies, and XY sex reversal. Am. J. Med. Genet. 2004, 125A, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Slee, J.; Goldblatt, J. Further evidence for a syndrome of “apple peel” intestinal atresia, ocular anomalies and microcephaly. Clin. Genet. 1996, 50, 260–262. [Google Scholar] [CrossRef]
- Van Bever, Y.; van Hest, L.; Wolfs, R.; Tibboel, D.; Hoonaard, T.L.v.D.; Gischler, S.J. Exclusion of aPAX6, FOXC1, PITX2, and MYCN mutation in another patient with apple peel intestinal atresia, ocular anomalies and microcephaly and review of the literature. Am. J. Med. Genet. Part A 2008, 146A, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Waters, F.M.; Lloyd, I.C.; Clayton-Smith, J. Apple peel atresia in association with bilateral colobomatous malformation of the optic nerve heads, dysmorphic features, and learning disability—A new syndrome? Ophthalmic Genet. 2000, 21, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Maroilley, T.; Tarailo-Graovac, M. Uncovering Missing Heritability in Rare Diseases. Genes 2019, 10, 275. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Söylemez, O.; Ozanturk, A.; Mourtzi, N.; Akle, S.; Jungreis, I.; Muller, J.; Cassa, C.A.; Brand, H.; Mokry, J.A.; et al. Evidence for secondary-variant genetic burden and non-random distribution across biological modules in a recessive ciliopathy. Nat. Genet. 2020, 52, 1145–1150. [Google Scholar] [CrossRef]
- Focșa, I.O.; Budișteanu, M.; Bălgrădean, M. Clinical and genetic heterogeneity of primary ciliopathies (Review). Int. J. Mol. Med. 2021, 48, 176. [Google Scholar] [CrossRef]
- Harper, L.; Michel, J.-L.; de Napoli-Cocci, S.; Aulagne, M.-B.; Maurel, A.; Mazouzi, S.; Ramful, D. One-step management of apple-peel atresia. Acta Chir. Belg. 2009, 109, 775–777. [Google Scholar] [CrossRef]
- McConnachie, D.J.; Stow, J.L.; Mallett, A.J. Ciliopathies and the Kidney: A Review. Am. J. Kidney Dis. 2021, 77, 410–419. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Variants in CENPF | c.1068+1G>A; p.Glu289Valfs*33 homozygous | c.5920dup; p.Thr1974Asnfs*9/ c.8991del; p.Ser2998Alafs*23 | c.892G>T; p.Glu298Ter/5 kb intragenic deletion |
| Country of origin | Lithuania | Australia | Poland |
| Age at last examination (gender) | Neonatal, died at 26 days (female) | 7 years (female) | 7 years (male) |
| Perinatal history | Preterm (34 weeks), SGA | Born at term | Born at term, SGA |
| OFC at birth | 25.8 cm, 3 cm < 3rd centile | 31 cm, 2 cm < 3rd centile | 26 cm, 6.5 cm < 3rd centile |
| Development | Not assessed | Normal | Delayed, especially speech; ADHD |
| Brain MRI examination | Brain cyst (colpocephaly), corpus callosum hypoplasia, primitive gyrosulcal pattern | Not performed. Only ultrasound examination | Colpocephaly |
| Ocular examination | Microphthalmia, sclerocornea | Peters anomaly type 1 | Microphthalmia, sclerocornea |
| Gastrointestinal anomalies | Duodenal atresia apple-peel type | Jejunal and ileal atresia | Jejunal atresia apple-peel type IIIb intestinal atresia |
| Kidneys | Hypoplasia | Borderline size 3rd centile | Hypoplasia, dysplasia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misceo, D.; Senaratne, L.D.S.; Mero, I.-L.; Sundaram, A.Y.M.; Bjørnstad, P.M.; Szczałuba, K.; Gasperowicz, P.; Kamien, B.; Nedregaard, B.; Holmgren, A.; et al. Novel Loss of Function Variants in CENPF Including a Large Intragenic Deletion in Patients with Strømme Syndrome. Genes 2023, 14, 1985. https://doi.org/10.3390/genes14111985
Misceo D, Senaratne LDS, Mero I-L, Sundaram AYM, Bjørnstad PM, Szczałuba K, Gasperowicz P, Kamien B, Nedregaard B, Holmgren A, et al. Novel Loss of Function Variants in CENPF Including a Large Intragenic Deletion in Patients with Strømme Syndrome. Genes. 2023; 14(11):1985. https://doi.org/10.3390/genes14111985
Chicago/Turabian StyleMisceo, Doriana, Lokuliyanage Dona Samudita Senaratne, Inger-Lise Mero, Arvind Y. M. Sundaram, Pål Marius Bjørnstad, Krzysztof Szczałuba, Piotr Gasperowicz, Benjamin Kamien, Bård Nedregaard, Asbjørn Holmgren, and et al. 2023. "Novel Loss of Function Variants in CENPF Including a Large Intragenic Deletion in Patients with Strømme Syndrome" Genes 14, no. 11: 1985. https://doi.org/10.3390/genes14111985