

Genotype–Phenotype Correlations in 293 Russian Patients with Causal Fabry Disease Variants
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selective Screening of Classical FD
2.2. Selective Screening of Patients with HCM
2.3. Genomic DNA Extraction
2.4. Sanger Sequencing
2.5. Next-Generation Sequencing
2.6. Bioinformatic Analysis
3. Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Number of Cases | Number of Families |
|---|---|---|
| c.680G>A (p.R227Q) | 17 | 2 |
| c.1156C>T (p.Q386*) | 10 | 1 |
| c.644A>G (p.N215S) | 10 | 4 |
| c.801G>A (p.M267I) | 10 | 1 |
| c.550T>G (p.Y184D) | 9 | 1 |
| c.679C>T (p.R227*) | 9 | 4 |
| c.161T>C (p.L54P) | 8 | 2 |
| c.334C>T (p.R112C) | 7 | 4 |
| c.658C>T (p.R220*) | 7 | 5 |
| c.717A>G (p.I239M) | 7 | 3 |
| c.782G>T (p.G261V) | 7 | 1 |
| c.101A>G (p.N34S) | 6 | 2 |
| c.901C>T (p.R301*) | 6 | 2 |
| c.902G>A (p.R301Q) | 5 | 3 |
| c.1021G>A (p.E341K) | 4 | 3 |
| c.128G>T (p.G43V) | 4 | 2 |
| c.195-1G>A | 4 | 1 |
| c.416A>G (p.N139S) | 4 | 1 |
| c.444T>G (p.S148R) | 4 | 1 |
| c.521G>A (p.C174R) | 4 | 1 |
| c.548-1G>C | 4 | 1 |
| c.718_719del (p.K240Efs*9) | 4 | 2 |
| c.723dup (p.I242Yfs*8) | 4 | 1 |
| c.786del (p.W262*) | 4 | 1 |
| c.902G>T (p.R301L) | 4 | 1 |
| c.1025G>A (p.R342Q) | 3 | 1 |
| c.1049del (p.A350Vfs*2) | 3 | 1 |
| c.128_132del (p.G43Afs*11) | 3 | 1 |
| c.146G>C (p.R49P) | 3 | 1 |
| c.227T>C (p.M76T) | 3 | 1 |
| c.269G>A (p.C90Y) | 3 | 2 |
| c.508G>T (p.D170H) | 3 | 1 |
| c.548-2A>G | 3 | 1 |
| c.551A>G (p.Y184C) | 3 | 1 |
| c.560T>A (p.M187K) | 3 | 1 |
| c.671A>G (p.N224S) | 3 | 1 |
| c.899T>C (p.L300P) | 3 | 1 |
| c.949del (p.I317Lfs*31) | 3 | 1 |
| c.95T>A (p.L32Q) | 3 | 1 |
| c.103G>A (p.G35R) | 2 | 1 |
| c.1066C>T (p.R356W) | 2 | 1 |
| c.1085_1098del (p.P362Hfs*8) | 2 | 1 |
| c.1121_1123del (p.K374_G375delinsR) | 2 | 1 |
| c.1134T>A (p.C378*) | 2 | 2 |
| c.1163T>A (p.L388H) | 2 | 1 |
| c.1197G>A (p.W399*) | 2 | 1 |
| c.1231G>C (p.G411R) | 2 | 1 |
| c.1277_1278del (p.K426Rfs*11) | 2 | 1 |
| c.1287_1288dup (p.*430Fext*1) | 2 | 1 |
| c.143A>G (p.E48G) | 2 | 1 |
| c.145C>G (p.R49G) | 2 | 1 |
| c.166T>A (p.C56S) | 2 | 1 |
| c.19G>T (p.E7*) | 2 | 1 |
| c.442_450del (p.S148_G150del) | 2 | 1 |
| c.447del (p.F149Lfs*16) | 2 | 1 |
| c.44C>A (p.A15E) | 2 | 1 |
| c.613C>A (p.P205T) | 2 | 1 |
| c.758T>C (p.I253T) | 2 | 1 |
| c.781G>T (p.G261C) | 2 | 1 |
| c.821G>A (p.G274D) | 2 | 1 |
| c.847C>T (p.Q283*) | 2 | 2 |
| c.889T>C (p.S297P) | 2 | 1 |
| c.996_999del (p.Q333Efs*14) | 2 | 2 |
| c.1024C>T (p.R342*) | 1 | 1 |
| c.1031T>C (p.L344P) | 1 | 1 |
| c.1033_1034del (p.S345Rfs*29) | 1 | 1 |
| c.1072_1074del (p.E358del) | 1 | 1 |
| c.1091C>G (p.S364C) | 1 | 1 |
| c.109G>A (p.A37T) | 1 | 1 |
| c.1139_1149del (p.P380Hfs*15) | 1 | 1 |
| c.1211G>A (p.R404K) | 1 | 1 |
| c.1228A>G (p.T410A) | 1 | 1 |
| c.127G>A (p.G43Ser) | 1 | 1 |
| c.187T>C (p.C63R) | 1 | 1 |
| c.203T>C (p.L68P) | 1 | 1 |
| c.375del (p.H125Qfs*5) | 1 | 1 |
| c.400T>G (Y134D) | 1 | 1 |
| c.496C>G (p.L166V) | 1 | 1 |
| c.503A>G (p.K168R0 | 1 | 1 |
| c.539_547 + 9del (p.L180_G183delinsC) | 1 | 1 |
| c.547G>A (p.G183S) | 1 | 1 |
| c.596T>C (p.V199A) | 1 | 1 |
| c.612G>C (p.W204C) | 1 | 1 |
| c.614C>G (p.P205R) | 1 | 1 |
| c.667T>C (p.C223R) | 1 | 1 |
| c.782del (p.G261Vfs*8) | 1 | 1 |
| c.804A>C (p.L268F) | 1 | 1 |
| c.818T>C (p.F273S) | 1 | 1 |
| c.831G>A (p.W277*) | 1 | 1 |
| c.834del (p.Q279Sfs*3) | 1 | 1 |
| c.835C>A (p.Q279K) | 1 | 1 |
| c.844A>C (p.T282P) | 1 | 1 |
| c.847C>A (p.Q283K) | 1 | 1 |
| c.859T>C (p.W287R) | 1 | 1 |
| c.869T>C (p.M290T) | 1 | 1 |
| c.895G>C (p.D299H) | 1 | 1 |
| c.897C>A (p.D299E) | 1 | 1 |
| c.908T>G (p.I303S) | 1 | 1 |
| c.920C>G (p.A307G) | 1 | 1 |
| c.946del (p.V316*) | 1 | 1 |
| c.971T>G (p.L324W) | 1 | 1 |
| c.981A>C (p.Q327H) | 1 | 1 |
| c.982G>C (p.G328R) | 1 | 1 |
| c.983G>C (p.G328A) | 1 | 1 |
References
- Nance, C.S.; Klein, C.J.; Banikazemi, M.; Dikman, S.H.; Phelps, R.G.; McArthur, J.C.; Rodriguez, M.; Desnick, R.J. Later-Onset Fabry Disease: An adult variant presenting with the cramp-fasciculation syndrome. Arch. Neurol. 2006, 63, 453–457. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Kotanko, P.; Kramar, R.; Devrnja, D.; Paschke, E.; Voigtla, T.; Auinger, M.; Demmelbauer, K.; Lorenz, M.; Hauser, A.-C.; Kofler, H.-J.D.; et al. Results of a Nationwide Screening for Anderson-Fabry Disease among Dialysis Patients. J. Am. Soc. Nephrol. 2004, 15, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, B.; Takenaka, T.; Teraguchi, H.; Tei, C.; Lee, P.; McKenna, W.J.; Elliott, P.M. Prevalence of Anderson-Fabry Disease in Male Patients with Late Onset Hypertrophic Cardiomyopathy. Circulation 2002, 105, 1407–1411. [Google Scholar] [CrossRef]
- Rolfs, A.; Böttcher, T.; Zschiesche, M.; Morris, P.; Winchester, B.; Bauer, P.; Walter, U.; Mix, E.; Löhr, M.; Harzer, K.; et al. Prevalence of Fabry disease in patients with cryptogenic stroke: A prospective study. Lancet 2005, 366, 1794–1796. [Google Scholar] [CrossRef] [PubMed]
- Sirrs, S.; Hollak, C.; Merkel, M.; Sechi, A.; Glamuzina, E.; Janssen, M.C.; Lachmann, R.; Langendonk, J.; Scarpelli, M.; Omran, T.B.; et al. The Frequencies of Different Inborn Errors of Metabolism in Adult Metabolic Centres: Report from the SSIEM Adult Metabolic Physicians Group. JIMD Rep. 2016, 27, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional Characterisation of Alpha-Galactosidase A Mutations as a Basis for a New Classification System in Fabry Disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef]
- Bishop, D.F.; Calhoun, D.H.; Bernstein, H.S.; Hantzopoulos, P.; Quinn, M.; Desnick, R.J. Human alpha-galactosidase A: Nucleotide sequence of a cDNA clone encoding the mature enzyme. Proc. Nat. Acad. Sci. USA 1986, 83, 4859–4863. [Google Scholar] [CrossRef]
- Fuller, M.; Mellett, N.; Hein, L.K.; Brooks, D.A.; Meikle, P.J. Absence of α-galactosidase cross-correction in Fabry heterozygote cultured skin fibroblasts. Mol. Genet. Metab. 2015, 114, 268–273. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef]
- Wang, R.Y.; Lelis, A.; Mirocha, J.; Wilcox, W.R. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Anesthesia Analg. 2007, 9, 34–45. [Google Scholar] [CrossRef]
- Schiffmann, R. Fabry disease. Pharmacol. Ther. 2009, 122, 65–77. [Google Scholar] [CrossRef]
- Clarke, J.T. Narrative review: Fabry disease. Ann. Intern. Med. 2007, 146, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.; Johnson, A.; Winchester, B. Synthesis of novel internal standards for the quantitative determination of plasma ceramide trihexoside in Fabry disease by tandem mass spectrometry. FEBS Lett. 2002, 515, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Smid, B.E.; van der Tol, L.; Biegstraaten, M.; Linthorst, G.E.; Hollak, C.E.M.; Poorthuis, B.J.H.M. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J. Med. Genet. 2015, 52, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B. Fabry disease: Recent advances in pathology, diagnosis, treatment and monitoring. Orphanet J. Rare Dis. 2009, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Clarke, J.T.R.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of Fabry disease: Changing pattern of causes of death in FOS—Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef]
- Nakao, S.; Kodama, C.; Takenaka, T.; Tanaka, A.; Yasumoto, Y.; Yoshida, A.; Kanzaki, T.; Enriquez, A.L.; Eng, C.M.; Tanaka, H.; et al. Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype1. Kidney Int. 2003, 64, 801–807. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD®): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Rombach, S.M.; Smid, B.E.; Bouwman, M.G.; Linthorst, G.E.; Dijkgraaf, M.G.W.; Hollak, C.E.M. Long term enzyme replacement therapy for Fabry disease: Effectiveness on kidney, heart and brain. Orphanet J. Rare Dis. 2013, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Arngrímsson, R.; Barbey, F.; Boks, L.; Cecchi, F.; Deegan, P.B.; Feldt-Rasmussen, U.; Geberhiwot, T.; Germain, D.P.; Hendriksz, C.; et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: The European Fabry Working Group consensus document. Orphanet J. Rare Dis. 2015, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Moiseev, S.; Fomin, V.; Savostyanov, K.; Pushkov, A.; Moiseev, A.; Svistunov, A.; Namazova-Baranova, L. The Prevalence and Clinical Features of Fabry Disease in Hemodialysis Patients: Russian Nationwide Fabry Dialysis Screening Program. Nephron 2019, 141, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Savostyanov, K.; Pushkov, A.; Zhanin, I.; Mazanova, N.; Trufanov, S.; Pakhomov, A.; Alexeeva, A.; Sladkov, D.; Asanov, A.; Fisenko, A. The prevalence of Fabry disease among 1009 unrelated patients with hypertrophic cardiomyopathy: A Russian nationwide screening program using NGS technology. Orphanet J. Rare Dis. 2022, 17, 199. [Google Scholar] [CrossRef] [PubMed]
- Namazova-Baranova, L.S.; Baranov, A.A.; Pushkov, A.A. Fabry disease in children: A federal screening programme in Russia. Eur. J. Pediatr. 2017, 176, 1385–1391. [Google Scholar] [CrossRef]
- Lukas, J.; Scalia, S.; Eichler, S.; Pockrandt, A.-M.; Dehn, N.; Cozma, C.; Giese, A.; Rolfs, A. Functional and Clinical Consequences of Novel α-Galactosidase A Mutations in Fabry Disease. Hum. Mutat. 2016, 37, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Duning, T.; Schelleckes, M.; Schmitz, B.; Stander, S.; Rolfs, A.; Brand, S.-M.; Brand, E. Multifocal White Matter Lesions Associated with the D313Y Mutation of the α-Galactosidase A Gene. PLoS ONE 2013, 8, e55565. [Google Scholar] [CrossRef]
- Namazova-Baranova, L.; Savostyanov, K.; Sukhozhenko, A.; Pushkov, A.; Mazanova, N.; Pakhomov, A.; Baranov, A. 14 novel mutations of GLA gene as a result of selective Fabry disease screening in Russian Federation. Mol. Genet. Metab. 2017, 120, S101. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Chong, K.-W.; Hsu, J.-H.; Yu, H.-C.; Shih, C.-C.; Huang, C.-H.; Lin, S.-J.; Chen, C.-H.; Chiang, C.-C.; Ho, H.-J.; et al. High Incidence of the Cardiac Variant of Fabry Disease Revealed by Newborn Screening in the Taiwan Chinese Population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef]
- van der Tol, L.; Smid, B.E.; Poorthuis, B.J.H.M.; Biegstraaten, M.; Deprez, R.H.L.; Linthorst, G.E.; Hollak, C.E.M. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 2013, 51, 1–9. [Google Scholar] [CrossRef]
- Ramaswami, U.; Whybra, C.; Parini, R.; Pintos-Morell, G.; Mehta, A.; Sunder-Plassmann, G.; Widmer, U.; Beck, M.; on Behalf of the Fos European Investigators. Clinical manifestations of Fabry disease in children: Data from the Fabry Outcome Survey. Acta Paediatr. 2006, 95, 86–92. [Google Scholar] [CrossRef]
- Kuzenkova, L.; Namazova-Baranova, L.S.; Podkletnova, V.; Gevorkyan, K.; Vashakmadze, N.D.; Savostyanov, K.V.; Studenikin, V.; Pushkov, S. Fabry Disease: Symptoms in Children and Teenagers. Curr. Pediatr. 2015, 14, 341. (In Russian) [Google Scholar] [CrossRef]
- Ellaway, C. Paediatric Fabry disease. Transl. Pediatr. 2016, 5, 37–42. [Google Scholar] [CrossRef] [PubMed]
- de Alencar, D.O.; Netto, C.; Ashton-Prolla, P.; Giugliani, R.; Ribeiro-Dos-Santos, Â.; Pereira, F.; Matte, U.; Santos, N.; Santos, S. Fabry disease: Evidence for a regional founder effect of the GLA gene mutation 30delG in Brazilian patients. Mol. Genet. Metab. Rep. 2014, 1, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.P.; Winchester, B.G.; Malcolm, S. Mutation analysis in patients with the typical form of Anderson—Fabry disease. Hum. Mol. Genet. 1993, 2, 1051–1053. [Google Scholar] [CrossRef]
- Meaney, C.; Blanch, L.C.; Morris, C. A nonsense mutation (R220X) in the α-galactosidase A gene detected in a female carrier of Fabry disease. Hum. Mol. Genet. 1994, 3, 1019–1020. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Morgante, E.; Russo, M.A.; Scopelliti, F.; Grande, C.; Verardo, R.; Franciosa, P.; Chimenti, C. Pathology and Function of Conduction Tissue in Fabry Disease Cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2015, 8, 799–805. [Google Scholar] [CrossRef]
- Citro, V.; Peña-García, J.; Den-Haan, H.; Pérez-Sánchez, H.; Del Prete, R.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Identification of an Allosteric Binding Site on Human Lysosomal Alpha-Galactosidase Opens the Way to New Pharmacological Chaperones for Fabry Disease. PLoS ONE 2016, 11, e0165463. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.; Mehta, A.; Gal, A. Genotype and phenotype in Fabry disease: Analysis of the Fabry Outcome Survey. Acta Paediatr. 2005, 94, 87–92. [Google Scholar] [CrossRef]
- Ashton-Prolla, P.; Tong, B.; Shabbeer, J.; Astrin, K.H.; Eng, C.M.; Desnick, R.J. Fabry disease: Twenty-two novel mutations in the α-galactosidase A gene and genotype/phenotype correlations in severely and mildly affected hemizygotes and heterozygotes. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2000, 48, 227–235. [Google Scholar]
- Savostyanov, K.; Pushkov, A.; Mazanova, N.; Pak, L.; Kuzenkova, L.; Podkletnova, T.; Pakhomov, A.; Sukhozhenko, A.; Moiseev, S. Lyso-GB3 is as a primary biomarker for Fabry disease screening among high-risk contingents. Mol. Genet. Metab. 2019, 126, S130. [Google Scholar] [CrossRef]
- Lavalle, L.; Thomas, A.S.; Beaton, B.; Ebrahim, H.; Reed, M.; Ramaswami, U.; Elliott, P.; Mehta, A.B.; Hughes, D.A. Phenotype and biochemical heterogeneity in late onset Fabry disease defined by N215S mutation. PLoS ONE 2018, 13, e0193550. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Rolfs, A.; Störk, S.; Bijnens, B.; Breunig, F.; Beer, M.; Ertl, G.; Wanner, C.; Weidemann, F. Gene Mutations Versus Clinically Relevant Phenotypes. Circ. Cardiovasc. Genet. 2014, 7, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, H.; Togawa, T.; Tsukimura, T.; Kato, H. Plasma lyso-Gb3: A biomarker for monitoring fabry patients during enzyme replacement therapy. Clin. Exp. Nephrol. 2017, 22, 843–849. [Google Scholar] [CrossRef] [PubMed]
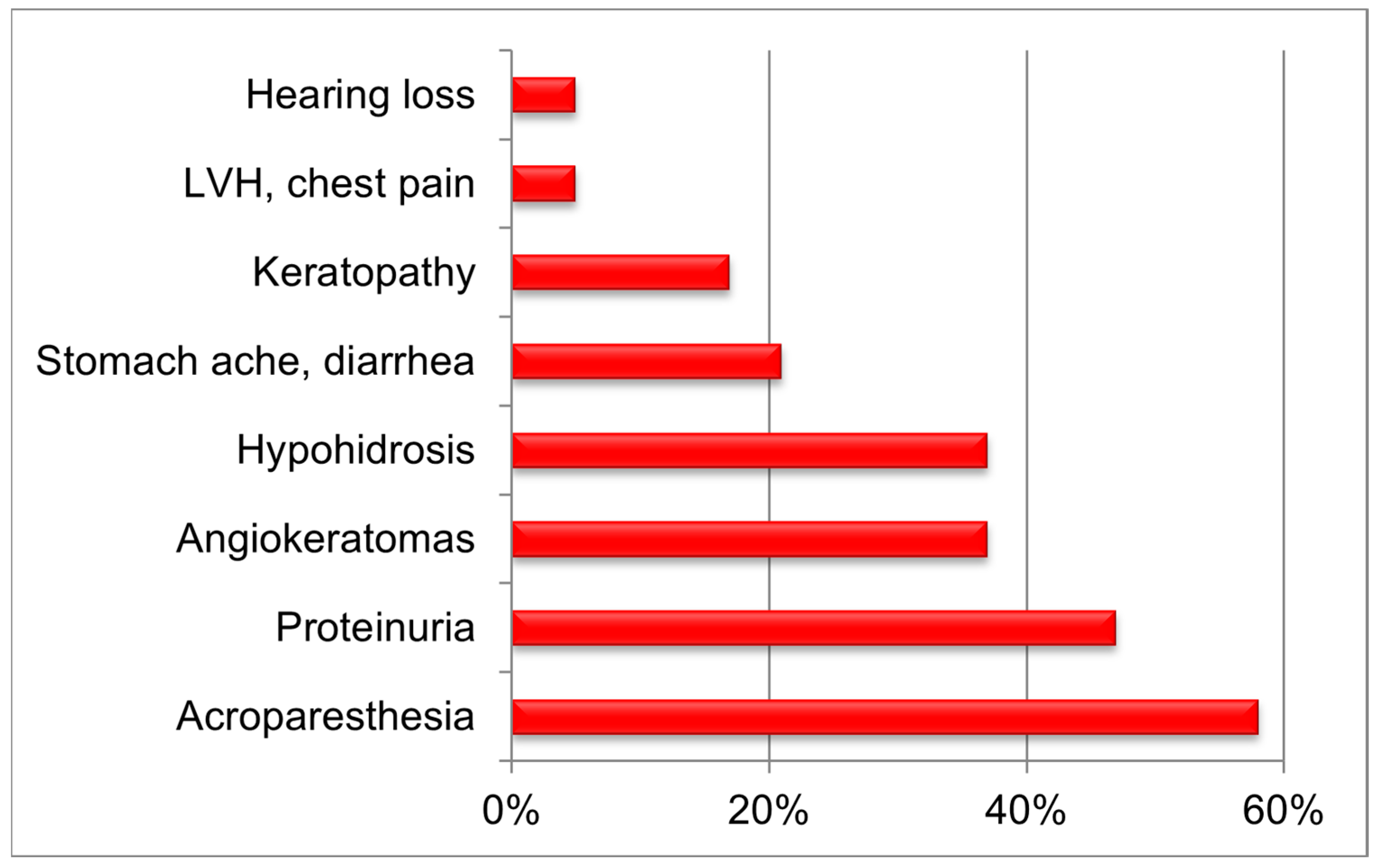



| Nucleotide Variant | Amino Acid Variant | Gender | Age of Diagnosis, Years | Lyso-Gb3, ng/mL | Keratopathy | Hypohidrosis | Acroparesthesia | Angiokeratoma | LVH, Chest Pain | Strokes | Proteinuria/Microalbuminuria | ESRD (Dialysis/Transplantation) | Hearing Loss | Stomachache/Vomiting/Diarrhea | Frequent Bronchitis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.203T>C | p.L68P | m | 52 | N/A | + | + | + | – | + | – | + | + | – | + | – |
| c.521G>A | p.C174Y | m | 18 | N/A | – | – | + | – | – | – | + | – | – | – | – |
| c.539_547 + 9del | p.L180_G183delinsC | m | 27 | 15.0 | – | + | + | + | – | – | – | – | – | + | – |
| c.804A>C | p.L268F | m | 19 | 75.9 | + | + | + | – | – | – | – | – | – | + | – |
| c.821G>A | p.G274D | m | 27 | 19.6 | + | + | + | + | – | – | + | + | – | + | + |
| c.844A>C | p.T282P | m | 30 | 17.3 | + | + | + | + | – | + | + | – | + | + | – |
| c.889T>C | p.S297P | m | 37 | 25.2 | N/A | + | + | + | – | – | + | – | – | + | – |
| c.895G>C | p.D299H | m | 68 | 9.95 | N/A | N/A | N/A | N/A | N/A | N/A | + | + | – | – | – |
| c.902G>T | p.R301L | f | 22 | 2.1 | – | – | – | – | – | – | – | – | – | – | – |
| c.981A>C | p.Q327H | f | 13 | 6.0 | – | + | + | – | – | – | + | – | – | – | – |
| c.1163T>A | p.L388H | m | 18 | N/A | + | + | + | + | – | – | + | – | – | – | – |
| c.949del | p.I317Lfs*31 | m | 21 | 60.2 | + | + | + | + | – | – | + | – | + | – | – |
| c.1134T>A | p.C378* | m | 28 | 73.1 | + | + | + | + | + | + | + | + | + | – | + |
| c.1211G>A | p.R404K | f | 15 | 0.8 | – | – | + | + | + | – | – | – | – | – | – |
| Indicator | “Quantitative” Variants | Other Variants | p-Value (Mann–Whitney U Test) |
|---|---|---|---|
| Lyso-Gb3 level, ng/mL | 17.9 (4.5–39.7) | 5.6 (2.8–24.0) | 0.003 |
| Age of diagnosis, years | 34.5 (23.5–48.0) | 36.5 (20.0–48.5) | 0.933 |
| Indicator | Male | Female | p-Value (Mann–Whitney U Test) |
|---|---|---|---|
| Lyso-Gb3 level, ng/mL | 30.1 (14.0–49.9) | 3.0 (1.6–4.9) | <0.001 |
| Age of diagnosis, years | 32.0 (18.0–45.0) | 40.5 (25.0–56.0) | <0.001 |
| Indicator | c.644A>G (p.N215S), c.717A>G (p.I239M), c.758T>C (p.I253T), c.895G>C (p.D299H) | Other Variants | p-Value (Mann–Whitney U Test) |
|---|---|---|---|
| Lyso-Gb3 level, ng/mL | 5.0 (4.1–8.6) | 31.1 (19.1–59.5) | <0.001 |
| Age of diagnosis, years | 41.0 (38.0–48.0) | 32.0 (18.0–45.0) | 0.073 |
| Indicator | Number of Patients with Stroke | Number of Patients without Stroke | p-Value (F-Test) |
|---|---|---|---|
| Other variants | 7 | 61 | 0.073 |
| “Quantitative” variants | 8 | 24 |
| Indicator | Number of Men with Stroke | Number of Men without Stroke | p-Value (F-Test) |
|---|---|---|---|
| Other variants | 3 | 39 | 0.005 |
| “Quantitative” variants | 7 | 11 |
| Indicator | Number of Patients with ESRD | Number of Patients without ESRD | p-Value (F-Test) |
|---|---|---|---|
| Other variants | 18 | 49 | 0.805 |
| “Quantitative” variants | 7 | 25 |
| Indicator | Patients with ESRD | Other Patients | p-Value (Mann–Whitney U Test) |
|---|---|---|---|
| Lyso-Gb3 level, ng/mL | 47.8 (19.6–66.3) | 17.6 (2.5–29.0) | 0.003 |
| Indicator | Patients with ESRD + Stroke | Other Patients | p-Value (Mann–Whitney U Test) |
|---|---|---|---|
| Lyso-Gb3 level, ng/mL | 95.5 (73.1–101.0) | 31.5 (21.0–98.0) | 0.004 |
| ERT | Number of Patients | Lyso-Gb3 Level, ng/mL | p-Value (Wilcoxon Test) |
|---|---|---|---|
| Before ERT | 54 | 39.3 (19.1–63.4) | <0.001 |
| During ERT | 12 | 21.3 (10.1–35.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savostyanov, K.; Pushkov, A.; Zhanin, I.; Mazanova, N.; Pakhomov, A.; Trufanova, E.; Alexeeva, A.; Sladkov, D.; Kuzenkova, L.; Asanov, A.; et al. Genotype–Phenotype Correlations in 293 Russian Patients with Causal Fabry Disease Variants. Genes 2023, 14, 2016. https://doi.org/10.3390/genes14112016
Savostyanov K, Pushkov A, Zhanin I, Mazanova N, Pakhomov A, Trufanova E, Alexeeva A, Sladkov D, Kuzenkova L, Asanov A, et al. Genotype–Phenotype Correlations in 293 Russian Patients with Causal Fabry Disease Variants. Genes. 2023; 14(11):2016. https://doi.org/10.3390/genes14112016
Chicago/Turabian StyleSavostyanov, Kirill, Alexander Pushkov, Ilya Zhanin, Natalya Mazanova, Alexander Pakhomov, Elena Trufanova, Alina Alexeeva, Dmitry Sladkov, Ludmila Kuzenkova, Aliy Asanov, and et al. 2023. "Genotype–Phenotype Correlations in 293 Russian Patients with Causal Fabry Disease Variants" Genes 14, no. 11: 2016. https://doi.org/10.3390/genes14112016
APA StyleSavostyanov, K., Pushkov, A., Zhanin, I., Mazanova, N., Pakhomov, A., Trufanova, E., Alexeeva, A., Sladkov, D., Kuzenkova, L., Asanov, A., & Fisenko, A. (2023). Genotype–Phenotype Correlations in 293 Russian Patients with Causal Fabry Disease Variants. Genes, 14(11), 2016. https://doi.org/10.3390/genes14112016