
Base-Excision Repair Mutational Signature in Two Sebaceous Carcinomas of the Eyelid
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
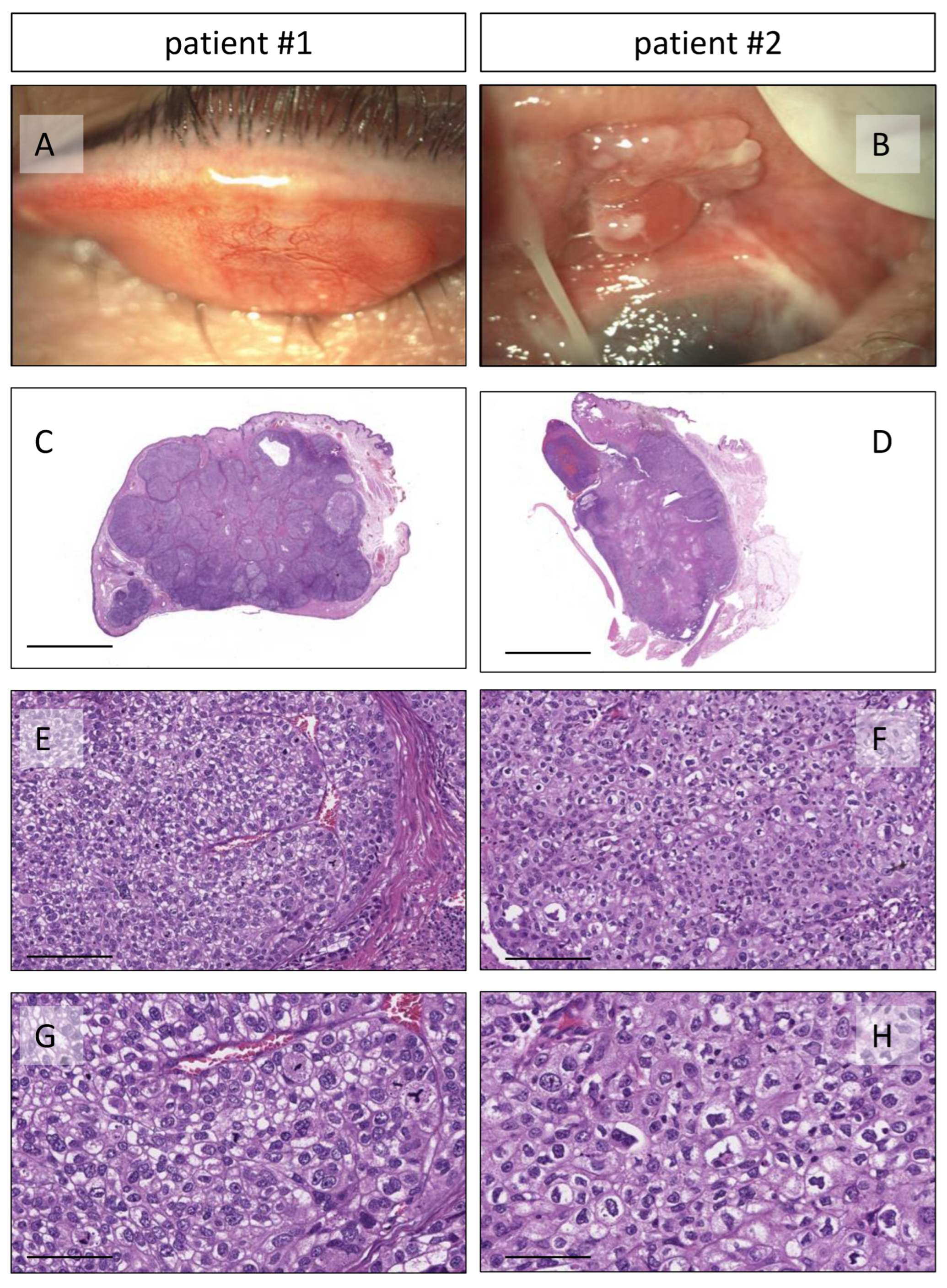
2.1. Patients
2.2. Sequencing
2.3. Bioinformatics Analyses
2.4. Microsatellite Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Orr, C.K.; Yazdanie, F.; Shinder, R. Current Review of Sebaceous Cell Carcinoma. Curr. Opin. Ophthalmol. 2018, 29, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Granada, C.; Rodriguez-Waitkus, P. Sebaceous Carcinoma of the Eyelid. Cancer Control 2016, 23, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Liang, W.-Y.; Tsai, C.-C.; Kao, S.-C.; Yu, W.-K.; Kau, H.-C.; Liu, C.J.-L. Comparison of the Clinical Characteristics and Outcome of Benign and Malignant Eyelid Tumors: An Analysis of 4521 Eyelid Tumors in a Tertiary Medical Center. BioMed Res. Int. 2015, 2015, 453091. [Google Scholar] [CrossRef]
- Alfaar, A.S.; Suckert, C.N.; Rehak, M.; Girbardt, C. The Epidemiology of Adults’ Eyelid Malignancies in Germany between 2009 and 2015: An Analysis of 42,710 Patients’ Data. Eur. J. Ophthalmol. 2022, 33, 1186–1199. [Google Scholar] [CrossRef]
- Tryggvason, G.; Bayon, R.; Pagedar, N.A. Epidemiology of Sebaceous Carcinoma of the Head and Neck: Implications for Lymph Node Management. Head Neck 2012, 34, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Sun, M.T.; Pirbhai, A.; Ueda, K.; Katori, N.; Selva, D. Sebaceous Carcinoma in Japanese Patients: Clinical Presentation, Staging and Outcomes. Br. J. Ophthalmol. 2013, 97, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, A.-S.; Campolmi, N.; Tumahai, P.; Kantelip, B.; Delbosc, B. Sebaceous Carcinoma of the Eyelid and Muir-Torre Syndrome. JAMA Ophthalmol. 2014, 132, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- Jagan, L.; Zoroquiain, P.; Bravo-Filho, V.; Logan, P.; Qutub, M.; Burnier, M.N. Sebaceous Adenomas of the Eyelid and Muir-Torre Syndrome. Br. J. Ophthalmol. 2015, 99, 909–913. [Google Scholar] [CrossRef]
- North, J.P.; Golovato, J.; Vaske, C.J.; Sanborn, J.Z.; Nguyen, A.; Wu, W.; Goode, B.; Stevers, M.; McMullen, K.; Perez White, B.E.; et al. Cell of Origin and Mutation Pattern Define Three Clinically Distinct Classes of Sebaceous Carcinoma. Nat. Commun. 2018, 9, 1894. [Google Scholar] [CrossRef]
- Tetzlaff, M.T.; Curry, J.L.; Ning, J.; Sagiv, O.; Kandl, T.L.; Peng, B.; Bell, D.; Routbort, M.; Hudgens, C.W.; Ivan, D.; et al. Distinct Biological Types of Ocular Adnexal Sebaceous Carcinoma: HPV-Driven and Virus-Negative Tumors Arise through Nonoverlapping Molecular-Genetic Alterations. Clin. Cancer Res. 2019, 25, 1280–1290. [Google Scholar] [CrossRef]
- Tetzlaff, M.T.; Singh, R.R.; Seviour, E.G.; Curry, J.L.; Hudgens, C.W.; Bell, D.; Wimmer, D.A.; Ning, J.; Czerniak, B.A.; Zhang, L.; et al. Next-Generation Sequencing Identifies High Frequency of Mutations in Potentially Clinically Actionable Genes in Sebaceous Carcinoma. J. Pathol. 2016, 240, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Hao, P.; Li, J.; Han, R.; Lin, J.; Li, X. Integrated Whole-Exome and Transcriptome Sequencing Indicated Dysregulation of Cholesterol Metabolism in Eyelid Sebaceous Gland Carcinoma. Transl. Vis. Sci. Technol. 2023, 12, 4. [Google Scholar] [CrossRef]
- Xu, S.; Moss, T.J.; Laura Rubin, M.; Ning, J.; Eterovic, K.; Yu, H.; Jia, R.; Fan, X.; Tetzlaff, M.T.; Esmaeli, B. Whole-Exome Sequencing for Ocular Adnexal Sebaceous Carcinoma Suggests PCDH15 as a Novel Mutation Associated with Metastasis. Mod. Pathol. 2020, 33, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- North, J.P.; Solomon, D.A.; Golovato, J.; Bloomer, M.; Benz, S.C.; Cho, R.J. Loss of ZNF750 in Ocular and Cutaneous Sebaceous Carcinoma. J. Cutan. Pathol. 2019, 46, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.L.; Kibbi, N.; Worley, B.; Kelm, R.C.; Wang, J.V.; Barker, C.A.; Behshad, R.; Bichakjian, C.K.; Bolotin, D.; Bordeaux, J.S.; et al. Sebaceous Carcinoma: Evidence-Based Clinical Practice Guidelines. Lancet Oncol. 2019, 20, e699–e714. [Google Scholar] [CrossRef] [PubMed]
- Płachta, I.; Kleibert, M.; Czarnecka, A.M.; Spałek, M.; Szumera-Ciećkiewicz, A.; Rutkowski, P. Current Diagnosis and Treatment Options for Cutaneous Adnexal Neoplasms with Follicular Differentiation. Int. J. Mol. Sci. 2021, 22, 4759. [Google Scholar] [CrossRef] [PubMed]
- Yin, V.T.; Merritt, H.A.; Sniegowski, M.; Esmaeli, B. Eyelid and Ocular Surface Carcinoma: Diagnosis and Management. Clin. Dermatol. 2015, 33, 159–169. [Google Scholar] [CrossRef]
- Goecks, J.; Nekrutenko, A.; Taylor, J.; Team, T.G. Galaxy: A Comprehensive Approach for Supporting Accessible, Reproducible, and Transparent Computational Research in the Life Sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; der Auwera, G.A.V.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. bioRxiv 2018. bioRxiv:201178. [Google Scholar]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Karlberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and Accurate Calling of Germline and Somatic Variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Larson, D.E.; Wilson, R.K. Using VarScan 2 for Germline Variant Calling and Somatic Mutation Detection. Curr. Protoc. Bioinform. 2013, 44, 15.4.1–15.4.17. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, K. Genomic Variant Annotation and Prioritization with ANNOVAR and WANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, A.; Amarante, T.D.; Czarnecki, J.; Shooter, S.; Zou, X.; Glodzik, D.; Morganella, S.; Nanda, A.S.; Badja, C.; Koh, G.; et al. A Practical Framework and Online Tool for Mutational Signature Analyses Show Inter-Tissue Variation and Driver Dependencies. Nat. Cancer 2020, 1, 249–263. [Google Scholar] [CrossRef]
- Lee, J.; Lee, A.J.; Lee, J.-K.; Park, J.; Kwon, Y.; Park, S.; Chun, H.; Ju, Y.S.; Hong, D. Mutalisk: A Web-Based Somatic MUTation AnaLyIS ToolKit for Genomic, Transcriptional and Epigenomic Signatures. Nucleic Acids Res. 2018, 46, W102–W108. [Google Scholar] [CrossRef]
- Díaz-Gay, M.; Vangara, R.; Barnes, M.; Wang, X.; Islam, S.M.A.; Vermes, I.; Narasimman, N.B.; Yang, T.; Jiang, Z.; Moody, S.; et al. Assigning Mutational Signatures to Individual Samples and Individual Somatic Mutations with SigProfilerAssignment. bioRxiv 2023. bioRxiv:2023.07.10.548264. [Google Scholar]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter Annotates the Biological and Clinical Relevance of Tumor Alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef]
- Stelzer, G.; Plaschkes, I.; Oz-Levi, D.; Alkelai, A.; Olender, T.; Zimmerman, S.; Twik, M.; Belinky, F.; Fishilevich, S.; Nudel, R.; et al. VarElect: The Phenotype-Based Variation Prioritizer of the GeneCards Suite. BMC Genom. 2016, 17 (Suppl. S2), 444. [Google Scholar] [CrossRef]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-Modified Human Stem Cell Organoids to Study the Origin of Mutational Signatures in Cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zethoven, M.; McInerny, S.; Devereux, L.; Huang, Y.-K.; Thio, N.; Cheasley, D.; Gutiérrez-Enríquez, S.; Moles-Fernández, A.; Diez, O.; et al. Evaluation of the Association of Heterozygous Germline Variants in NTHL1 with Breast Cancer Predisposition: An International Multi-Center Study of 47,180 Subjects. NPJ Breast Cancer 2021, 7, 52. [Google Scholar] [CrossRef]
- Santen, G.W.E.; Aten, E.; Sun, Y.; Almomani, R.; Gilissen, C.; Nielsen, M.; Kant, S.G.; Snoeck, I.N.; Peeters, E.A.J.; Hilhorst-Hofstee, Y.; et al. Mutations in SWI/SNF Chromatin Remodeling Complex Gene ARID1B Cause Coffin-Siris Syndrome. Nat. Genet. 2012, 44, 379–380. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Zhu, W.; Xu, Y.; Ma, J.; He, C.; Wang, F. SWI/SNF Complex Gene Variations Are Associated with a Higher Tumor Mutational Burden and a Better Response to Immune Checkpoint Inhibitor Treatment: A Pan-Cancer Analysis of next-Generation Sequencing Data Corresponding to 4591 Cases. Cancer Cell Int. 2022, 22, 347. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Lalloo, F.; Ryan, N.A.; Bowers, N.; Green, K.; Woodward, E.R.; Clancy, T.; Bolton, J.; McVey, R.J.; Wallace, A.J.; et al. Advances in Genetic Technologies Result in Improved Diagnosis of Mismatch Repair Deficiency in Colorectal and Endometrial Cancers. J. Med. Genet. 2022, 59, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Keshinro, A.; Ganesh, K.; Vanderbilt, C.; Firat, C.; Kim, J.K.; Chen, C.-T.; Yaeger, R.; Segal, N.H.; Gonen, M.; Shia, J.; et al. Characteristics of Mismatch Repair-Deficient Colon Cancer in Relation to Mismatch Repair Protein Loss, Hypermethylation Silencing, and Constitutional and Biallelic Somatic Mismatch Repair Gene Pathogenic Variants. Dis. Colon Rectum 2023, 66, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]

{kind=link}
| Mutational Signatures from Signal | ||||
|---|---|---|---|---|
| Patient #1 | SBS30 54% BER deficiency | SBS1 28% Deamination | SBS18 16% BER deficiency | Unassigned 1% |
| Patient #2 | SBS30 68% BER deficiency | SBS1 30% Deamination | Unassigned 2% | |
| Top 5 Most Similar Cancer Samples from Signal | |||||
|---|---|---|---|---|---|
| Patient #1 | GEL-2944131-11 CNS 0.877 | GEL-2478942-11 CNS 0.797 | GEL-2808113-11 Bone and soft tissues 0.761 | DO52610 Bone and soft tissues 0.748 | DO52610 NET 0.74 |
| Patient #2 | GEL-2944131-11 CNS 0.917 | GEL-2478942-11 CNS 0.856 | GEL-2808113-11 Bone and soft tissues 0.827 | DO52610 Bone and soft tissues 0.807 | HMF0030688 Bone and soft tissues 0.796 |
| Mutational Signatures from Mutalisk Patient #1 | |||
|---|---|---|---|
| Signatures | Probabilities | Cosine | BIC |
| 23 30 1 7 22 18 | 0.27861 0.26564 0.17885 0.13563 0.07967 0.06159 | 0.82600 | 17,428.410 |
| 30 23 1 7 11 22 18 | 0.24343 0.23671 0.18389 0.10866 0.08411 0.08106 0.06216 | 0.82700 | 17,435.600 |
| 30 23 1 7 22 | 0.30204 0.27441 0.20025 0.13004 0.09325 | 0.82300 | 17,654.570 |
| 30 23 1 7 | 0.40833 0.26234 0.22351 0.10581 | 0.81500 | 17,970.830 |
| 30 23 1 | 0.54416 0.25502 0.20081 | 0.80800 | 18,044.040 |
| 1 | 1.00000 | 0.74800 | 19,951.940 |
| 30 23 | 0.75283 0.24717 | 0.78800 | 21,139.930 |
| Mutational signatures from Mutalisk Patient #2 | |||
| Signatures | Probabilities | Cosine | BIC |
| 30 23 1 7 11 22 4 | 0.30286 0.26799 0.20011 0.11531 0.07099 0.03274 0.01000 | 0.90100 | 23,356.170 |
| 30 23 1 7 11 22 | 0.30818 0.26830 0.20242 0.11439 0.07076 0.03594 | 0.90100 | 23,392.800 |
| 30 23 1 7 22 | 0.32650 0.30361 0.19804 0.13716 0.03469 | 0.90000 | 23,397.800 |
| 30 23 1 7 | 0.36591 0.29917 0.20674 0.12819 | 0.89900 | 23,550.040 |
| 30 23 1 | 0.53017 0.29046 0.17937 | 0.89100 | 23,718.120 |
| 30 23 | 0.71638 0.28362 | 0.87200 | 26,641.600 |
| 19 | 1.0000 | 0.82400 | 26,767.810 |
| Patient | Mutation | Gene | Protein Change | CGI-Oncogenic Summary | Consequence | LOH |
|---|---|---|---|---|---|---|
| #1 | chr10:89717672 C>T | PTEN | R233X | oncogenic (predicted and annotated) | stop_gained | YES |
| #1 | chr17:7578419 C>A | TP53 | E171X | oncogenic (predicted) | stop_gained | NO |
| #1 | chr17:80790252-80790252 ATTT>- | ZNF750 | KC26-27X | oncogenic (predicted) | frameshift_variant | NO |
| #1 | chr22:37708082 C>T | CYTH4 | P327S | oncogenic (predicted) | missense_variant | NO |
| #1 | chr2:141116501 G>A | LRP1B | P3716S | oncogenic (predicted) | missense_variant | NO |
| #1 | chr3:52610685 G>A | PBRM1 | A1188V | oncogenic (predicted) | missense_variant | NO |
| #1 | chr4:54319218 C>T | FIP1L1 | R473C | oncogenic (predicted) | missense_variant | NO |
| #1 | chr4:187557927 G>A | FAT1 | R1262X | oncogenic (predicted) | stop_gained | YES |
| #1 | chr6:157502150 C>G | ARID1B | Y1184X | oncogenic (predicted) | stop_gained | NO |
| #1 | chr7:143098557 C>T | EPHA1 | E98K | oncogenic (predicted) | missense_variant | NO |
| #2 | chr13:48951171 G>T | RB1 | -- | oncogenic (predicted and annotated) | splice_donor_variant | NO |
| #2 | chr12:992225 G>A | WNK1 | M1576I | oncogenic (predicted) | missense_variant | NO |
| #2 | chr12:56482607 C>A | ERBB3 | T355N | oncogenic (predicted) | missense_variant | NO |
| #2 | chr13:35734171 T>A | NBEA | -- | oncogenic (predicted) | splice_donor_variant | YES |
| #2 | chr13:35756496 G>T | NBEA | -- | oncogenic (predicted) | splice_acceptor_variant | YES |
| #2 | chr14:21875095 G>A | CHD8 | R943C | oncogenic (predicted) | missense_variant | NO |
| #2 | chr15:28231765 C>T | OCA2 | E403K | oncogenic (predicted) | missense_variant | YXES |
| #2 | chr16:58585089 C>T | CNOT1 | E1097K | oncogenic (predicted) | missense_variant | NO |
| #2 | chr17:1561923 G>A | PRPF8 | P1758L | oncogenic (predicted) | missense_variant | YES |
| #2 | chr17:7800428 C>T | CHD3 | R638X | oncogenic (predicted) | stop_gained | YES |
| #2 | chr17:8050603 C>T | PER1 | G532R | oncogenic (predicted) | missense_variant | NO |
| #2 | chr17:56438147-56438147 C>- | RNF43 | G282X | oncogenic (predicted) | frameshift_variant | NO |
| #2 | chr17:62496853 G>A | DDX5 | P419S | oncogenic (predicted) | missense_variant | YES |
| #2 | chr18:50912508 G>A | DCC | D819N | oncogenic (predicted) | missense_variant | YES |
| #2 | chr19:9084065 C>A | MUC16 | G2584X | oncogenic (predicted) | stop_gained | YES |
| #2 | chr1:21190980 C>T | EIF4G3 | E1132K | oncogenic (predicted) | missense_variant | NO |
| #2 | chr1:155920771 C>T | ARHGEF2 | R994H | oncogenic (predicted) | missense_variant | NO |
| #2 | chr1:197091163 G>A | ASPM | T1251I | oncogenic (predicted) | missense_variant | NO |
| #2 | chr22:20074748 C>T | DGCR8 | R262X | oncogenic (predicted) | stop_gained | YES |
| #2 | chr3:13672957 G>A | FBLN2 | D1072N | oncogenic (predicted) | missense_variant | NO |
| #2 | chr3:71050163 C>G | FOXP1 | C341S | oncogenic (predicted) | missense_variant | NO |
| #2 | chr3:185146526 C>T | MAP3K13 | R53X | oncogenic (predicted) | stop_gained | NO |
| #2 | chr6:94068121 G>A | EPHA7 | R281C | oncogenic (predicted) | missense_variant | YES |
| #2 | chr6:157517340 C>A | ARID1B | P1425T | oncogenic (predicted) | missense_variant | YES |
| #2 | chr8:31024573 C>T | WRN | P1340S | oncogenic (predicted) | missense_variant | YES |
| #2 | chr8:103340036 C>T | UBR5 | R472Q | oncogenic (predicted) | missense_variant | NO |
| #2 | chr9:8633420 T>A | PTPRD | R83S | oncogenic (predicted) | missense_variant | NO |
| #2 | chrX:44923062 G>A | KDM6A | Q693 | oncogenic (predicted) | splice_region_variant | YES |
| Patient | Top 20 Genes with LOH | Gene Name |
|---|---|---|
| #1 | BRCA2 | BRCA2 DNA Repair-Associated |
| #1 | BRIP1 | BRCA1 Interacting Helicase 1 |
| #1 | NF1 | Neurofibromin 1 |
| #1 | BARD1 | BRCA1 Associated RING Domain 1 |
| #1 | PTEN | Phosphatase and Tensin Homolog |
| #1 | AXIN2 | Axin 2 |
| #1 | STK11 | Serine/Threonine Kinase 11 |
| #1 | RET | Ret Proto-Oncogene |
| #1 | RAD51C | RAD51 Paralog C |
| #1 | KIT | KIT Proto-Oncogene, Receptor Tyrosine Kinase |
| #1 | BMPR1A | Bone Morphogenetic Protein Receptor Type 1A |
| #1 | FLCN | Folliculin |
| #1 | ERBB2 | Erb-B2 Receptor Tyrosine Kinase 2 |
| #1 | FANCC | FA Complementation Group C |
| #1 | SUFU | SUFU Negative Regulator of Hedgehog Signaling |
| #1 | GALNT12 | Polypeptide N-Acetylgalactosaminyltransferase 12 |
| #1 | PRKAR1A | Protein Kinase CAMP-Dependent Type I Regulatory Subunit α |
| #1 | STAT3 | Signal Transducer and Activator of Transcription 3 |
| #1 | ERBB3 | Erb-B2 Receptor Tyrosine Kinase 3 |
| #1 | LRRC56 | Leucine Rich Repeat Containing 56 |
| #2 | BRCA1 | BRCA1 DNA Repair Associated |
| #2 | ATM | ATM Serine/Threonine Kinase |
| #2 | PALB2 | Partner and Localizer of BRCA2 |
| #2 | MSH6 | MutS Homolog 6 |
| #2 | MSH2 | MutS Homolog 2 |
| #2 | CDH1 | Cadherin 1 |
| #2 | BRIP1 | BRCA1 Interacting Helicase 1 |
| #2 | PMS2 | PMS1 Homolog 2, Mismatch Repair System Component |
| #2 | POLE | DNA Polymerase Epsilon, Catalytic Subunit |
| #2 | NF1 | Neurofibromin 1 |
| #2 | TSC2 | TSC Complex Subunit 2 |
| #2 | ALK | ALK Receptor Tyrosine Kinase |
| #2 | DICER1 | Dicer 1, Ribonuclease III |
| #2 | RET | Ret Proto-Oncogene |
| #2 | NTHL1 | Nth Like DNA Glycosylase 1 |
| #2 | MLH3 | MutL Homolog 3 |
| #2 | ERBB2 | Erb-B2 Receptor Tyrosine Kinase 2 |
| #2 | PALLD | Palladin, Cytoskeletal-Associated Protein |
| #2 | WT1 | WT1 Transcription Factor |
| #2 | AOPEP | Aminopeptidase O (Putative) |
| Patient | Gene | Variant | Consequence |
|---|---|---|---|
| #1 | ALDH1A1 | chr9:75543807 C>T | splice_donor_variant |
| #1 | ABCA1 | chr2:215884469 T>A; p.(Ser447Cys) | missense_variant |
| #1 | CAGE1 | chr6:7370306 C>A | splice_region_variant |
| #1 | RUBCNL | chr13:46918890 C>T; p.(Cys621Tyr) | missense_variant |
| #1 | COL4A2 | chr13:111156539 G>T; p.(Gly1444Cys) | missense_variant |
| #1 | PLCE1 | chr10:96081817 G>A | splice_donor_variant |
| #1 | SYNE1 | chr6:152683380 C>T; p.(Met3408Ile) | missense_variant |
| #1 | MADCAM1 | chr19:501786 C>A; p.(Pro286Gln) | missense_variant |
| #1 | NET1 | chr10:5498201 G>A; p.(Gly450Asp) | missense_variant |
| #1 | HSPG2 | chr1:22170691 C>T; p.(Ala2856Thr) | missense_variant |
| #2 | MSH6 | chr2:48027853 C>T; p.(Arg911ter) | stop_gained |
| #2 | ERBB2 | chr17:37881572 C>T | splice_region_variant |
| #2 | PALLD | chr4:169825046 G>A; p.(Ala871Thr) | missense_variant |
| #2 | FANCD2 | chr3:10106408 C>T | splice_region_variant |
| #2 | KMT2C | chr7:151970859 C>T; p.(Gly315Ser) | missense_variant |
| #2 | TTN | chr2:179584566 G>A | splice_region_variant |
| #2 | FASN | chr17:80041523 G>A | splice_region_variant |
| #2 | LIG1 | chr19:48665601 A>T; p.(Phe9Ile) | missense_variant |
| #2 | DSPP | chr4:88533649 G>A; p.(Gly104Glu) | missense_variant |
| #2 | ITGA6 | chr2:173330387 C>G; p.(Asn101Lys) | missense_variant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sangiorgi, E.; Giannuzzi, F.; Molinario, C.; Rapari, G.; Riccio, M.; Cuffaro, G.; Castri, F.; Benvenuto, R.; Genuardi, M.; Massi, D.; et al. Base-Excision Repair Mutational Signature in Two Sebaceous Carcinomas of the Eyelid. Genes 2023, 14, 2055. https://doi.org/10.3390/genes14112055
Sangiorgi E, Giannuzzi F, Molinario C, Rapari G, Riccio M, Cuffaro G, Castri F, Benvenuto R, Genuardi M, Massi D, et al. Base-Excision Repair Mutational Signature in Two Sebaceous Carcinomas of the Eyelid. Genes. 2023; 14(11):2055. https://doi.org/10.3390/genes14112055
Chicago/Turabian StyleSangiorgi, Eugenio, Federico Giannuzzi, Clelia Molinario, Giulia Rapari, Melania Riccio, Giovanni Cuffaro, Federica Castri, Roberta Benvenuto, Maurizio Genuardi, Daniela Massi, and et al. 2023. "Base-Excision Repair Mutational Signature in Two Sebaceous Carcinomas of the Eyelid" Genes 14, no. 11: 2055. https://doi.org/10.3390/genes14112055