
Genetic Spectrum and Cascade Screening of Familial Hypercholesterolemia in Routine Clinical Setting in Hong Kong

 ,
, Highlights
- The majority of the causative variants identified in individuals with Familial Hypercholesterolemia (FH) in Hong Kong were in the LDLR gene.
- The currently used clinical criteria identified genetically confirmed FH cases with low specificity, but the treatment intensity required to achieve a significant reduction in LDL-C levels may serve as an alternative tool.
- Genetic cascade screening has proven valuable in detecting genetically confirmed FH in family members who may exhibit less severe phenotypes that might otherwise go undetected.
- These findings underscore the significance of genetic testing and cascade screening in the accurate identification and management of FH.
Abstract
:1. Introduction

{kind=link}
{kind=link}
| Criteria | Simon Broome Register | MEDPED | DLCNC | Modified DLCNC | JFHMC | Hong Kong Expert Panel Recommendation |
|---|---|---|---|---|---|---|
| Family history of pCVD or hyperlipidemia | Y | Y | Y | Y | Y | Optional |
| History of pCVD | N | N | Y | Y | N | N |
| Physical signs (e.g., tendon xanthoma) | Y | N | Y | Y | Y | Optional |
| LDL-C cutoff (mmol/L) | Adult: >4.9 Children: > 4.0 | By total cholesterol: specific levels based on individual’s age and a family history of FH | • ≥8.5: 8 points • 6.5–8.4: 5 points • 5.0–6.4: 3 points • 4.0–4.9: 1 point | • ≥6: 8 points • 5–5.9: 5 points • 3.5–4.9: 3 points • 2.5–3.4: 1 point | For HeFH: Adult: >4.7 Children: >3.6 For HoFH: Total cholesterol: >15.5 mmol/L | Adult: >5; >4.5 if with family history of FH or pCVD Children: >3.6 if with family history; >4.9 and/or physical signs (e.g., xanthomata) |
| Genetic study | Optional | N | Optional | Optional | N | Optional |
| Diagnosis | • Definite FH • Possible FH | With FH | • Definite FH • Probable FH • Possible FH | • Definite FH • Probable FH • Possible FH | With FH | With FH |
| Merit | • Higher specificity than MEDPED • Ease of remembrance • Economic viability | • Higher sensitivity than DLCNC and Simon Broome Register criteria • Easy to use | • Higher specificity than MEDPED • Each criterion is weighted • Molecular defect leading to FH addressed | • Higher sensitivity than DLCNC | • High specificity and sensitivity in Japanese population | • Not evaluated so far |
| Demerit | • Lower sensitivity than MEDPED • Cannot discriminate between FH and secondary causes | • Lower specificity than DLCNC and Simon Broome Register criteria • Without regard to physical symptoms and a history of pCVD | • Lower sensitivity than MEDPED • Not applicable to children • Lack of versatility of use | • Lower specificity than DLCNC |
2. Materials and Methods
3. Results
3.1. Genetic Spectrum Identified in the Entire Cohort
3.2. Findings in the Adult Cohort
3.3. Findings in the Pediatric Cohort
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marks, D.; Thorogood, M.; Neil, H.A.; Humphries, S.E. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003, 168, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Benn, M.; Watts, G.F.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Familial hypercholesterolemia in the danish general population: Prevalence, coronary artery disease, and cholesterol-lowering medication. J. Clin. Endocrinol. Metab. 2012, 97, 3956–3964. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Charng, M.J. Genetic Diagnosis of Familial Hypercholesterolemia in Asia. Front. Genet. 2020, 11, 833. [Google Scholar] [CrossRef]
- Pang, J.; Chan, D.C.; Hu, M.; Muir, L.A.; Kwok, S.; Charng, M.-J.; Florkowski, C.M.; George, P.M.; Lin, J.; Loi, D.D.; et al. Comparative aspects of the care of familial hypercholesterolemia in the “Ten Countries Study”. J. Clin. Lipidol. 2019, 13, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Hu, M.; Lin, J.; Miida, T.; Nawawi, H.M.; Park, J.E.; Wu, X.; Ramli, A.S.; Kim, N.T.; Kwok, S.; et al. An enquiry based on a standardised questionnaire into knowledge, awareness and preferences concerning the care of familial hypercholesterolaemia among primary care physicians in the Asia-Pacific region: The “Ten Countries Study”. BMJ Open 2017, 7, e017817. [Google Scholar] [CrossRef]
- Representatives of the Global Familial Hypercholesterolemia Community; Wilemon, K.A.; Patel, J.; Ahmed, C.D.; Alkhnifsawi, M.; Almahmeed, W.; Alonso, R.; Al-Rasadi, K.; Badimon, L.; Bernal, L.M.; et al. Reducing the Clinical and Public Health Burden of Familial Hypercholesterolemia: A Global Call to Action. JAMA Cardiol. 2020, 5, 217–229. [Google Scholar] [CrossRef]
- Tomlinson, B.; Chan, J.C.; Chan, W.B.; Chen, W.W.; Chow, F.C.; Li, S.K.; Kong, A.P.; Ma, R.C.; Siu, D.C.; Tan, K.C.; et al. Guidance on the management of familial hypercholesterolaemia in Hong Kong: An expert panel consensus viewpoin. Hong Kong Med. J. 2018, 24, 408–415. [Google Scholar] [CrossRef]
- Chiou, K.R.; Charng, M.J. Common mutations of familial hypercholesterolemia patients in Taiwan: Characteristics and implications of migrations from southeast China. Gene 2012, 498, 100–106. [Google Scholar] [CrossRef]
- Jiang, L.; Sun, L.Y.; Dai, Y.F.; Yang, S.W.; Zhang, F.; Wang, L.Y. The distribution and characteristics of LDL receptor mutations in China: A systematic review. Sci. Rep. 2015, 5, 17272. [Google Scholar] [CrossRef]
- Chiou, K.R.; Charng, M.J. Genetic diagnosis of familial hypercholesterolemia in Han Chinese. J. Clin. Lipidol. 2016, 10, 490–496. [Google Scholar] [CrossRef]
- Sun, D.; Zhou, B.Y.; Li, S.; Sun, N.-L.; Hua, Q.; Wu, S.-L.; Cao, Y.-S.; Guo, Y.-L.; Wu, N.-Q.; Zhu, C.-G.; et al. Genetic basis of index patients with familial hypercholesterolemia in Chinese population: Mutation spectrum and genotype-phenotype correlation. Lipids Health Dis. 2018, 17, 252. [Google Scholar] [CrossRef]
- Meng, R.; Wei, Q.; Zhou, J.; Zhang, B.; Li, C.; Shen, M. A systematic review of cost-effectiveness analysis of different screening strategies for familial hypercholesterolemia. J. Clin. Lipidol. 2023, 17, 1–8. [Google Scholar] [CrossRef]
- Al-Rasadi, K.; Al-Waili, K.; Al-Sabti, H.A.; Al-Hinai, A.; Al-Hashmi, K.; Al-Zakwani, I.; Banerjee, Y. Criteria for Diagnosis of Familial Hypercholesterolemia: A Comprehensive Analysis of the Different Guidelines, Appraising their Suitability in the Omani Arab Population. Oman Med. J. 2014, 29, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yuan, B.; Zhao, D.; Taylor, A.W.; Lin, J.; Watts, G.F. Familial hypercholesterolemia in China: Prevalence and evidence of underdetection and undertreatment in a community population. Int. J. Cardiol. 2014, 174, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Cheung, C.L.; Yeung, C.Y.; Siu, D.; Leung, J.; Pang, H.K. Genetic screening for familial hypercholesterolaemia in Hong Kong. Hong Kong Med. J. 2018, 24, 7–10. [Google Scholar] [PubMed]
- Haralambos, K.; Ashfield-Watt, P.; McDowell, I.F. Diagnostic scoring for familial hypercholesterolaemia in practice. Curr. Opin. Lipidol. 2016, 27, 367–374. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Ison, H.E.; Clarke, S.L.; Knowles, J.W. Familial hypercholesterolemia, GeneReviews® [Internet]. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK174884/ (accessed on 10 November 2023).
- Turkyilmaz, A.; Kurnaz, E.; Alavanda, C.; Yarali, O.; Baykan, E.K.; Yavuz, D.; Cayir, A.; Ata, P. The Spectrum of Low-Density Lipoprotein Receptor Mutations in a Large Turkish Cohort of Patients with Familial Hypercholesterolemia. Metab. Syndr. Relat. Disord. 2021, 19, 340–346. [Google Scholar] [CrossRef]
- Chan, M.L.; Cheung, C.L.; Lee, A.C.; Yeung, C.; Siu, C.; Leung, J.Y.; Pang, H.; Tan, K.C. Genetic variations in familial hypercholesterolemia and cascade screening in East Asians. Mol. Genet. Genom. Med. 2019, 7, e00520. [Google Scholar] [CrossRef] [PubMed]
- Mak, Y.T.; Pang, C.P.; Tomlinson, B.; Zhang, J.; Chan, Y.-S.; Mak, T.W.L.; Masarei, J.R.L. Mutations in the low-density lipoprotein receptor gene in Chinese familial hypercholesterolemia patients. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1600–1605. [Google Scholar] [CrossRef]
- Hu, M.; Hooper, A.J.; Bockxmeer, F.M.; Watts, G.F.; Chan, J.C.; Tomlinson, B. Management of Familial Hypercholesterolemia in Hong Kong. J. Atheroscler. Thromb. 2016, 23, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Sturm, A.; Knowles, J.; Gidding, S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef]
- Gazzotti, M.; Olmastroni, E.; Pederiva, C.; Capra, M.; Catapano, A.; Casula, M. Diagnostic criteria in children and adolescents affected by familial hypercholesterolemia in the Italian lipigen paediatric group. Atherosclerosis 2021, 331, e49. [Google Scholar] [CrossRef]
- Khera, A.V.; Won, H.H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef]
- Mandraffino, G.; Scicali, R.; Rodríguez-Carrio, J.; Savarino, F.; Mamone, F.; Scuruchi, M.; Cinquegrani, M.; Imbalzano, E.; Di Pino, A.; Piro, S.; et al. Arterial stiffness improvement after adding on PCSK9 inhibitors or ezetimibe to high-intensity statins in patients with familial hypercholesterolemia: A Two-Lipid Center Real-World Experience. J. Clin. Lipidol. 2020, 14, 231–240. [Google Scholar] [CrossRef]
- Pang, J.; Chan, D.C.; Watts, G.F. The Knowns and Unknowns of Contemporary Statin Therapy for Familial Hypercholesterolemia. Curr. Atheroscler. Rep. 2020, 22, 64. [Google Scholar] [CrossRef]
- Liao, J.K. Safety and efficacy of statins in Asians. Am. J. Cardiol. 2007, 99, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar]
- Aparicio, A.; Villazón, F.; Suárez-Gutiérrez, L.; Gómez, J.; Martínez-Faedo, C.; Méndez-Torre, E.; Avanzas, P.; Álvarez-Velasco, R.; Cuesta-Llavona, E.; García-Lago, C.; et al. Clinical Evaluation of Patients with Genetically Confirmed Familial Hypercholesterolemia. J. Clin. Med. 2023, 12, 1030. [Google Scholar] [CrossRef]
- Alonso, R.; Perez de Isla, L.; Muñiz-Grijalvo, O.; Mata, P. Barriers to Early Diagnosis and Treatment of Familial Hypercholesterolemia: Current Perspectives on Improving Patient Care. Vasc. Health Risk Manag. 2020, 16, 11–25. [Google Scholar] [CrossRef]
- Medeiros, A.M.; Bourbon, M. Genetic Testing in Familial Hypercholesterolemia: Is It for Everyone? Curr. Atheroscler. Rep. 2023, 25, 127–132. [Google Scholar] [CrossRef]
- McGowan, M.P.; Cuchel, M.; Ahmed, C.D.; Khera, A.; Weintraub, W.S.; Wilemon, K.A.; Ahmad, Z. A proof-of-concept study of cascade screening for Familial Hypercholesterolemia in the US, adapted from the Dutch model. Am. J. Prev. Cardiol. 2021, 6, 100170. [Google Scholar] [CrossRef]
- Lázaro, P.; Pérez de Isla, L.; Watts, G.F.; Alonso, R.; Norman, R.; Muñiz, O.; Fuentes, F.; Mata, N.; López-Miranda, J.; González-Juanatey, J.R.; et al. Cost-effectiveness of a cascade screening program for the early detection of familial hypercholesterolemia. J. Clin. Lipidol. 2017, 11, 260–271. [Google Scholar] [CrossRef]
- Wonderling, D.; Umans-Eckenhausen, M.A.; Marks, D.; Defesche, J.C.; Kastelein, J.J.; Thorogood, M. Cost-effectiveness analysis of the genetic screening program for familial hypercholesterolemia in The Netherlands. Semin. Vasc. Med. 2004, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ban, M.R.; Hegele, R.A. Multiplex ligation-dependent probe amplification of LDLR enhances molecular diagnosis of familial hypercholesterolemia. J. Lipid. Res. 2005, 46, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, M.; Tichý, L.; Freiberger, T.; Blaha, V.; Satny, M.; Hubacek, J.A. Genetics of Familial Hypercholesterolemia: New Insights. Front. Genet. 2020, 11, 574474. [Google Scholar] [CrossRef] [PubMed]
- Chiou, K.R.; Charng, M.J. Detection of mutations and large rearrangements of the low-density lipoprotein receptor gene in Taiwanese patients with familial hypercholesterolemia. Am. J. Cardiol. 2010, 105, 1752–1758. [Google Scholar] [CrossRef]
- Salen, G.; Patel, S.; Batta, A.K. Sitosterolemia. Cardiovasc. Drug Rev. 2002, 20, 255–270. [Google Scholar] [CrossRef]
- Kao, J.T.; Wen, H.C.; Chien, K.L.; Hsu, H.C.; Lin, S.W. A novel genetic variant in the apolipoprotein A5 gene is associated with hypertriglyceridemia. Hum. Mol. Genet. 2003, 12, 2533–2539. [Google Scholar] [CrossRef]
- Fawwad, A.; Sabir, R.; Riaz, M.; Moin, H.; Basit, A. Measured versus calculated LDL-cholesterol in subjects with type 2 diabetes. Pak. J. Med. Sci. 2016, 32, 955–960. [Google Scholar] [CrossRef]

| Types of LDLR Variants | Total Number of Cases | Adult | Pediatric | Total Number of Unique Variants | Newly Reported in Hong Kong Population | Total Number of Novel Variants |
|---|---|---|---|---|---|---|
| All | 22 | 16 | 6 | 25 | 14 | 2 |
| (1) Single variant | 18 | 14 | 4 | 17 | 10 | 1 |
| Receptor-negative | 6 | 5 | 1 | 7 | 6 | 1 |
| Splicing | 3 | 3 | 0 | 3 | 3 | 1 |
| Frameshift | 1 | 1 | 0 | 1 | 1 | 0 |
| Nonsense | 2 | 1 | 1 | 2 | 2 | 0 |
| Receptor-defective | 12 | 9 | 3 | 10 | 4 | 0 |
| Missense | 12 | 9 | 3 | 10 | 4 | 0 |
| (2) Two variants | 4 | 2 | 2 | 8 | 4 | 1 |
| Defective + Negative | 2 | 0 | 2 | 4 | 3 | 1 |
| Defective + Defective | 2 | 2 | 0 | 4 | 1 | 0 |
| Variant | Protein Change | Localization | No. of Probands (No. by Cascade Screening) | Clinical Significance |
|---|---|---|---|---|
| LDLR NM_000527.5: | ||||
| c.268 G>A | p.(Asp90Asn) | 3 | 1 | Pathogenic |
| c.301 G>A | p.(Glu101Lys) | 3 | 1 | Pathogenic |
| c.523 G>A | p.(Asp175Asn) | 4 | 1 | Pathogenic |
| c.769 C>T | p.(Arg257Trp) | 5 | 1 | VUS |
| c.837 dupC | p.(Asn280GlnfsTer21) # | 6 | 1 | Likely pathogenic |
| c.986 G>A | p.(Cys329Tyr) | 7 | 2 (1) | Likely pathogenic |
| c.1055 G>A | p.(Cys352Tyr) | 7 | 1 | Pathogenic |
| c.1060+2 T>C | Splice donor variant # | Intron 7 | 1 | Pathogenic |
| c.1216 C>A | p.(Arg406=) New splice acceptor introduced | 9 | 1 | Likely pathogenic |
| c.1241 T>G | p.(Leu414Arg) | 9 | 3 (3) | Likely pathogenic |
| c.1247 G>A | p.(Arg416Gln) | 9 | 1 (1) | Pathogenic |
| c.1285 G>A | p.(Val429Met) | 9 | 1 | Pathogenic |
| c.1297 G>C | p.(Asp433His) | 9 | 1 | Likely pathogenic |
| c.1448 G>A | p.(Trp483Ter) | 10 | 1 (2) | Pathogenic |
| c.1469 G>A | p.(Trp490Ter) | 10 | 1 | Pathogenic |
| c.1586+5 G>C | Intron variant | Intron 10 | 1 | Likely pathogenic |
| c.1706-1 G>C | Splice acceptor variant | Intron 11 | 1 | Pathogenic |
| c.1731 G>C | p.(Trp577Cys) | 12 | 1 (4) | Pathogenic |
| c.1765 G>A | p.(Asp589Asn) | 12 | 1 (1) | VUS/Likely pathogenic |
| c.1880 C>T | p.(Ala627Val) | 13 | 1 (1) | Likely pathogenic |
| c.2001_2002 del | p.(Cys667*) | 13 | 1 | Pathogenic |
| c.2026 G>A | p.(Gly676Ser) | 13 | 1 | Likely pathogenic |
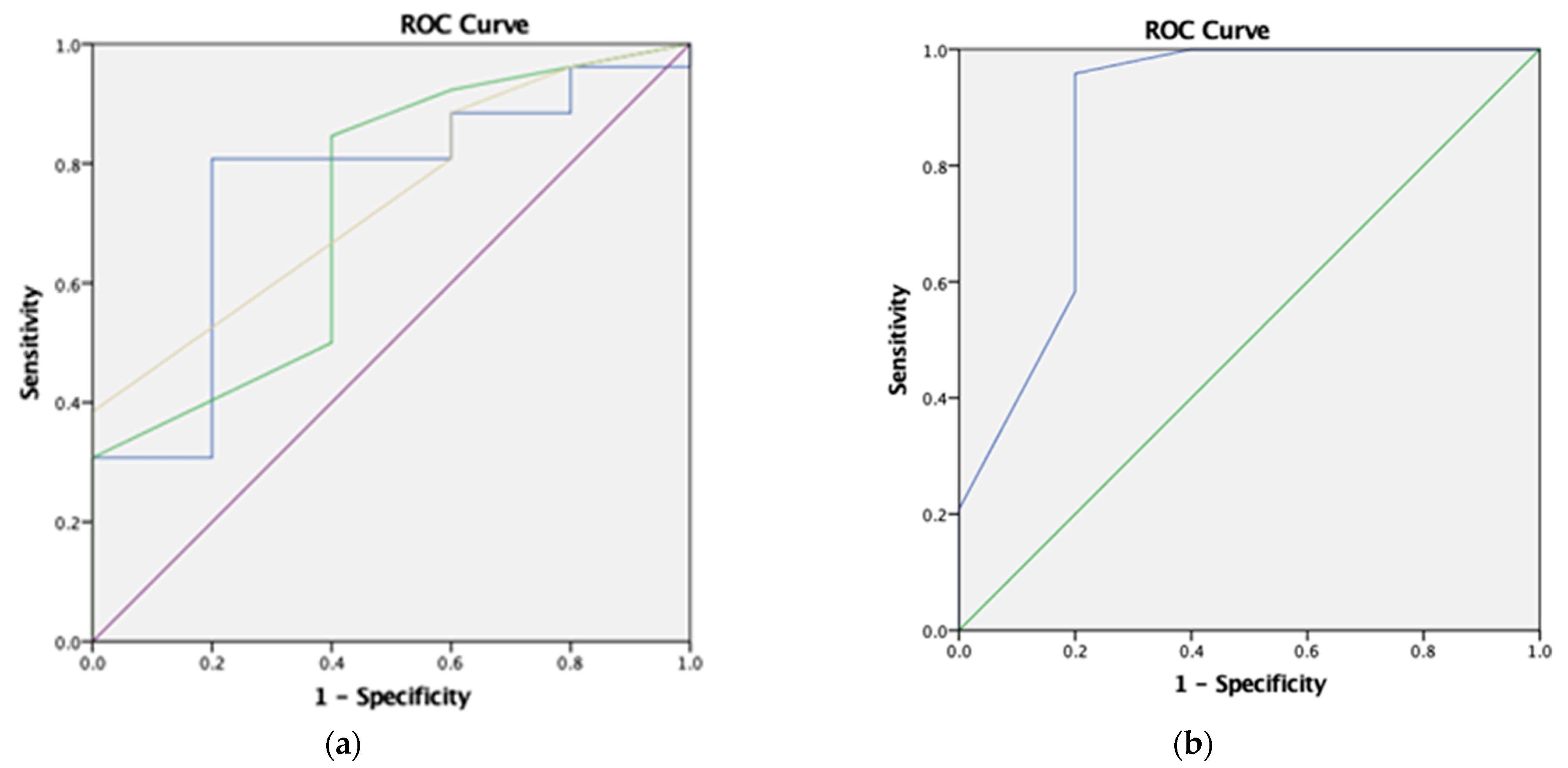
| Criteria (Prevalence = 84.2%) | Sensitivity | Specificity | PPV | NPV | Accuracy | p-Value |
|---|---|---|---|---|---|---|
| (1) Simon Broome Register | 84.6% | 40.0% | 88.0% | 33.3% | 77.4% | 0.241 |
| (2) MEDPED | 84.6% | 40.0% | 88.0% | 33.3% | 77.4% | 0.241 |
| (3) JFHMC | 84.6% | 40.0% | 88.0% | 33.3% | 77.4% | 0.241 |
| (4) Hong Kong guideline | 88.5% | 20.0% | 85.2% | 25.0% | 77.4% | 0.525 |
| (5) Pretreatment LDL-C ≥ 5.5 mmol/L | 80.8% | 60.0% | 91.3% | 37.5% | 77.4% | 0.093 |
| (6) Treatment intensity ≥ 1.8 to achieve a drop of pretreatment LDL-C by around 50% | 95.8% | 80.0% | 95.8% | 80.0% | 93.1% | 0.001 * |
| (7) Items 5 and 6 | 83.3% | 100.0% | 100.0% | 55.6% | 86.2% | 0.001 * |
| Criteria | Article | Sensitivity | Specificity |
|---|---|---|---|
| (1) Simon Broome Register | This study | 84.6% | 40.0% |
| [15] | 64.0% | 56.6% | |
| (2) DLCNC (cutoff of 3) | This study | 100.0% | 0.0% |
| [15] | 82.8% | 53.3% | |
| (3) Modified DLCNC (cutoff of 3) | This study | 100.0% | 0.0% |
| [15] | 93.8% | 26.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yip, M.-K.; Kwan, E.Y.-W.; Leung, J.Y.-Y.; Lau, E.Y.-F.; Poon, W.-T. Genetic Spectrum and Cascade Screening of Familial Hypercholesterolemia in Routine Clinical Setting in Hong Kong. Genes 2023, 14, 2071. https://doi.org/10.3390/genes14112071
Yip M-K, Kwan EY-W, Leung JY-Y, Lau EY-F, Poon W-T. Genetic Spectrum and Cascade Screening of Familial Hypercholesterolemia in Routine Clinical Setting in Hong Kong. Genes. 2023; 14(11):2071. https://doi.org/10.3390/genes14112071
Chicago/Turabian StyleYip, Man-Kwan, Elaine Yin-Wah Kwan, Jenny Yin-Yan Leung, Emmy Yuen-Fun Lau, and Wing-Tat Poon. 2023. "Genetic Spectrum and Cascade Screening of Familial Hypercholesterolemia in Routine Clinical Setting in Hong Kong" Genes 14, no. 11: 2071. https://doi.org/10.3390/genes14112071
APA StyleYip, M.-K., Kwan, E. Y.-W., Leung, J. Y.-Y., Lau, E. Y.-F., & Poon, W.-T. (2023). Genetic Spectrum and Cascade Screening of Familial Hypercholesterolemia in Routine Clinical Setting in Hong Kong. Genes, 14(11), 2071. https://doi.org/10.3390/genes14112071