
Unraveling the Genetic Basis of Feed Efficiency in Cattle through Integrated DNA Methylation and CattleGTEx Analysis
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cows and Determination of RFI
2.2. DNA Methylation and Data Processing
2.3. Associated CpG Site for RFI Identification
2.4. GO Enrichment Analysis
2.5. Co-Expression Using Expression Data from CattleGTEx
3. Results
3.1. Experimental Design
3.2. Methylation Data Generation and Summarization
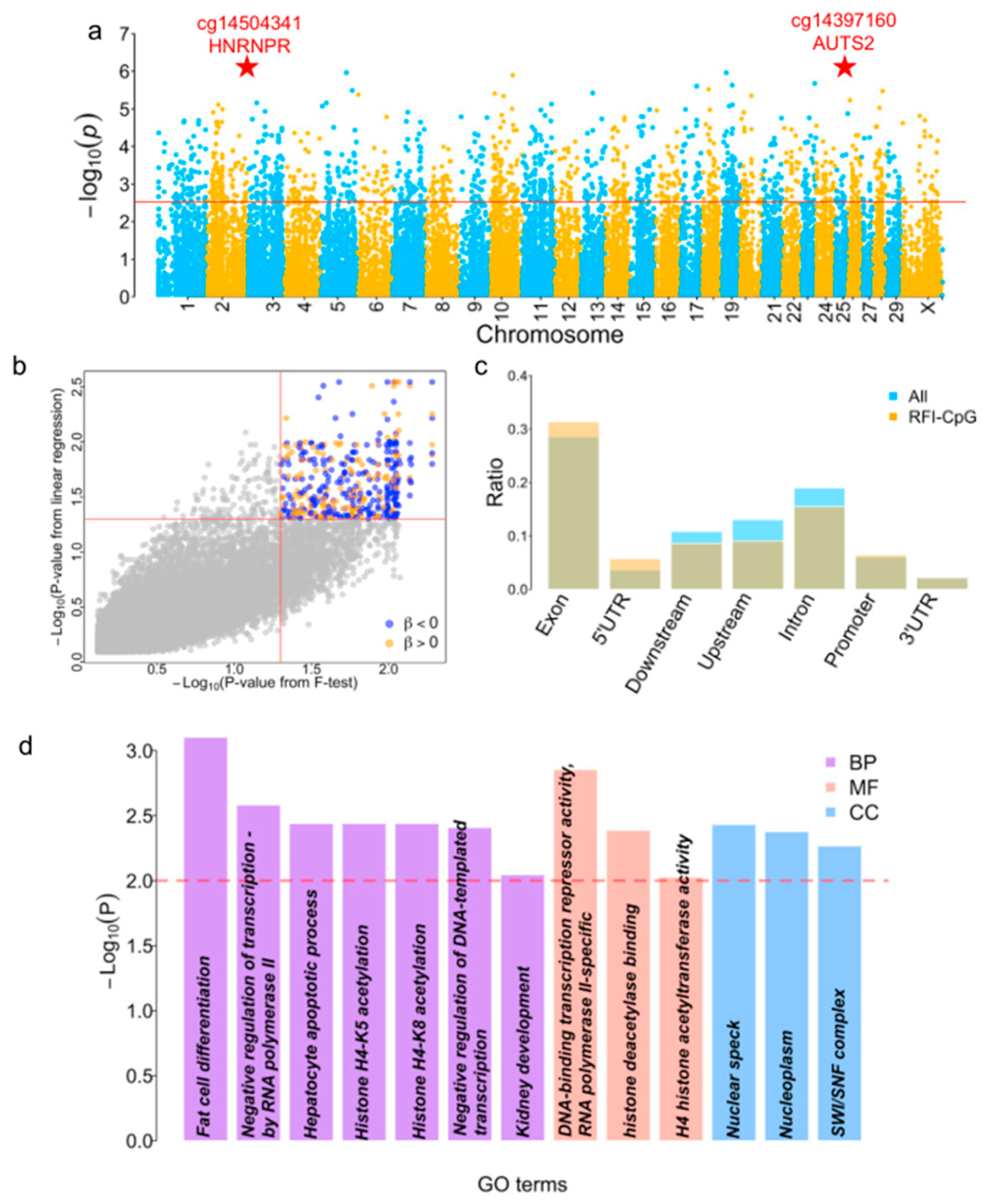
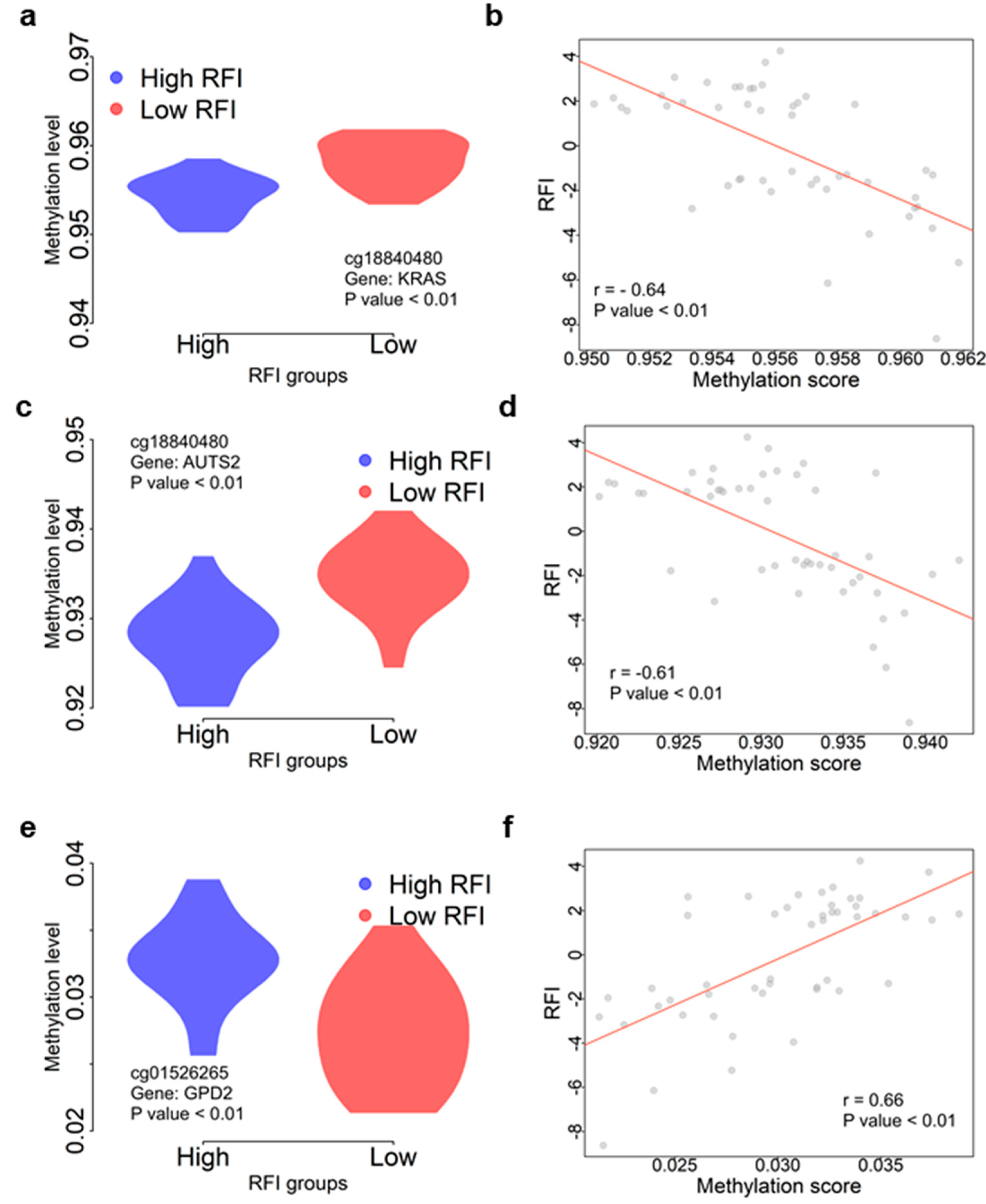
3.3. Identification of Differential Methylation CpG Sites between High and Low RFI Groups
3.4. Function Annotation of the FE Genes
3.5. Co-Expression Network of FE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilbert, M.; Nicolas, G.; Cinardi, G.; Van Boeckel, T.P.; Vanwambeke, S.O.; Wint, G.R.W.; Robinson, T.P. Global Distribution Data for Cattle, Buffaloes, Horses, Sheep, Goats, Pigs, Chickens and Ducks in 2010. Sci. Data 2018, 5, 180227. [Google Scholar] [CrossRef]
- Basarab, J.A.; Price, M.A.; Aalhus, J.L.; Okine, E.K.; Snelling, W.M.; Lyle, K.L. Residual Feed Intake and Body Composition in Young Growing Cattle. Can. J. Anim. Sci. 2003, 83, 189–204. [Google Scholar] [CrossRef]
- Kristiansen, S.; Painter, J.; Shea, M. Animal Agriculture and Climate Change in the US and UK Elite Media: Volume, Responsibilities, Causes and Solutions. Environ. Commun. 2021, 15, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Barthelmie, R.J. Impact of Dietary Meat and Animal Products on GHG Footprints: The UK and the US. Climate 2022, 10, 43. [Google Scholar] [CrossRef]
- Williams, Y.J.; Pryce, J.E.; Grainger, C.; Wales, W.J.; Linden, N.; Porker, M.; Hayes, B.J. Variation in Residual Feed Intake in Holstein-Friesian Dairy Heifers in Southern Australia. J. Dairy Sci. 2011, 94, 4715–4725. [Google Scholar] [CrossRef]
- Cavani, L.; Brown, W.E.; Parker Gaddis, K.L.; Tempelman, R.J.; VandeHaar, M.J.; White, H.M.; Peñagaricano, F.; Weigel, K.A. Estimates of Genetic Parameters for Feeding Behavior Traits and Their Associations with Feed Efficiency in Holstein Cows. J. Dairy Sci. 2022, 105, 7564–7574. [Google Scholar] [CrossRef] [PubMed]
- Khanal, P.; Johnson, J.; Gouveia, G.; Ross, P.; Deeb, N. Genomic Evaluation of Feed Efficiency in US Holstein Heifers. J. Dairy Sci. 2023, 106, 6986–6994. [Google Scholar] [CrossRef]
- Connor, E.E.; Hutchison, J.L.; Olson, K.M.; Norman, H.D. Triennial Lactation Symposium: Opportunities for Improving Milk Production Efficiency in Dairy Cattle. J. Anim. Sci. 2012, 90, 1687–1694. [Google Scholar] [CrossRef]
- Xi, Y.M.; Wu, F.; Zhao, D.Q.; Yang, Z.; Li, L.; Han, Z.Y.; Wang, G.L. Biological Mechanisms Related to Differences in Residual Feed Intake in Dairy Cows. Anim. Int. J. Anim. Biosci. 2016, 10, 1311–1318. [Google Scholar] [CrossRef]
- Nehme Marinho, M.; Zimpel, R.; Peñagaricano, F.; Santos, J.E.P. Assessing Feed Efficiency in Early and Mid Lactation and Its Associations with Performance and Health in Holstein Cows. J. Dairy Sci. 2021, 104, 5493–5507. [Google Scholar] [CrossRef]
- Elolimy, A.A.; Arroyo, J.M.; Batistel, F.; Iakiviak, M.A.; Loor, J.J. Association of Residual Feed Intake with Abundance of Ruminal Bacteria and Biopolymer Hydrolyzing Enzyme Activities during the Peripartal Period and Early Lactation in Holstein Dairy Cows. J. Anim. Sci. Biotechnol. 2018, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Fang, L.; Null, D.J.; Hutchison, J.L.; Connor, E.E.; VanRaden, P.M.; VandeHaar, M.J.; Tempelman, R.J.; Weigel, K.A.; Cole, J.B. High-Density Genome-Wide Association Study for Residual Feed Intake in Holstein Dairy Cattle. J. Dairy Sci. 2019, 102, 11067–11080. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; VanRaden, P.M.; Guduk, E.; O’Connell, J.R.; Null, D.J.; Connor, E.E.; VandeHaar, M.J.; Tempelman, R.J.; Weigel, K.A.; Cole, J.B. Genomic Prediction of Residual Feed Intake in US Holstein Dairy Cattle. J. Dairy Sci. 2020, 103, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kadarmideen, H.N. Metabolomics Analyses in High-Low Feed Efficient Dairy Cows Reveal Novel Biochemical Mechanisms and Predictive Biomarkers. Metabolites 2019, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Miglior, F.; Fonseca, P.A.S.; Gómez-Redondo, I.; Zeidan, J.; Suárez-Vega, A.; Schenkel, F.; Guan, L.L.; Waters, S.; Stothard, P.; et al. Identification of Functional Candidate Variants and Genes for Feed Efficiency in Holstein and Jersey Cattle Breeds Using RNA-Sequencing. J. Dairy Sci. 2021, 104, 1928–1950. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, H.F.; Zhou, Z.; Gomes, M.S.; Peixoto, P.M.G.; Bonsaglia, E.C.R.; Canisso, I.F.; Weimer, B.C.; Lima, F.S. Rumen and Lower Gut Microbiomes Relationship with Feed Efficiency and Production Traits throughout the Lactation of Holstein Dairy Cows. Sci. Rep. 2022, 12, 4904. [Google Scholar] [CrossRef] [PubMed]
- Freetly, H.C.; Kuehn, L.A.; Thallman, R.M.; Snelling, W.M. Heritability and Genetic Correlations of Feed Intake, Body Weight Gain, Residual Gain, and Residual Feed Intake of Beef Cattle as Heifers and Cows. J. Anim. Sci. 2020, 98, skz394. [Google Scholar] [CrossRef]
- Grünberg, W.; Staufenbiel, R.; Constable, P.D.; Dann, H.M.; Morin, D.E.; Drackley, J.K. Liver Phosphorus Content in Holstein-Friesian Cows during the Transition Period. J. Dairy Sci. 2009, 92, 2106–2117. [Google Scholar] [CrossRef]
- Moran, B.; Cummins, S.B.; Creevey, C.J.; Butler, S.T. Transcriptomics of Liver and Muscle in Holstein Cows Genetically Divergent for Fertility Highlight Differences in Nutrient Partitioning and Inflammation Processes. BMC Genom. 2016, 17, 603. [Google Scholar] [CrossRef]
- Pascottini, O.B.; De Koster, J.; Van Nieuwerburgh, F.; Van Poucke, M.; Peelman, L.; Fievez, V.; Leroy, J.L.M.R.; Opsomer, G. Effect of Overconditioning on the Hepatic Global Gene Expression Pattern of Dairy Cows at the End of Pregnancy. J. Dairy Sci. 2021, 104, 8152–8163. [Google Scholar] [CrossRef]
- Veshkini, A.; Hammon, H.M.; Vogel, L.; Delosière, M.; Viala, D.; Dèjean, S.; Tröscher, A.; Ceciliani, F.; Sauerwein, H.; Bonnet, M. Liver Proteome Profiling in Dairy Cows during the Transition from Gestation to Lactation: Effects of Supplementation with Essential Fatty Acids and Conjugated Linoleic Acids as Explored by PLS-DA. J. Proteom. 2022, 252, 104436. [Google Scholar] [CrossRef] [PubMed]
- Accorsi, P.A.; Pacioni, B.; Pezzi, C.; Forni, M.; Flint, D.J.; Seren, E. Role of Prolactin, Growth Hormone and Insulin-like Growth Factor 1 in Mammary Gland Involution in the Dairy Cow. J. Dairy Sci. 2002, 85, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Annen, E.L.; Fitzgerald, A.C.; Gentry, P.C.; McGuire, M.A.; Capuco, A.V.; Baumgard, L.H.; Collier, R.J. Effect of Continuous Milking and Bovine Somatotropin Supplementation on Mammary Epithelial Cell Turnover. J. Dairy Sci. 2007, 90, 165–183. [Google Scholar] [CrossRef]
- Bernier-Dodier, P.; Girard, C.L.; Talbot, B.G.; Lacasse, P. Effect of Dry Period Management on Mammary Gland Function and Its Endocrine Regulation in Dairy Cows. J. Dairy Sci. 2011, 94, 4922–4936. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Hata, E.; Sláma, P.; Kimura, K.; Hirai, T. Characteristics of Mammary Secretions from Holstein Cows at Approximately 10 Days before Parturition: With or without Intramammary Infection. J. Appl. Anim. Res. 2018, 46, 604–608. [Google Scholar] [CrossRef]
- Zhao, X.; Ponchon, B.; Lanctôt, S.; Lacasse, P. Invited Review: Accelerating Mammary Gland Involution after Drying-off in Dairy Cattle. J. Dairy Sci. 2019, 102, 6701–6717. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.; Terré, M.; Vidal, M. Symposium Review: Decomposing Efficiency of Milk Production and Maximizing Profit. J. Dairy Sci. 2020, 103, 5709–5725. [Google Scholar] [CrossRef]
- Aschenbach, J.R.; Zebeli, Q.; Patra, A.K.; Greco, G.; Amasheh, S.; Penner, G.B. Symposium Review: The Importance of the Ruminal Epithelial Barrier for a Healthy and Productive Cow. J. Dairy Sci. 2019, 102, 1866–1882. [Google Scholar] [CrossRef]
- Li, F.; Hitch, T.C.A.; Chen, Y.; Creevey, C.J.; Guan, L.L. Comparative Metagenomic and Metatranscriptomic Analyses Reveal the Breed Effect on the Rumen Microbiome and Its Associations with Feed Efficiency in Beef Cattle. Microbiome 2019, 7, 6. [Google Scholar] [CrossRef]
- Martin, M.J.; Pralle, R.S.; Bernstein, I.R.; VandeHaar, M.J.; Weigel, K.A.; Zhou, Z.; White, H.M. Circulating Metabolites Indicate Differences in High and Low Residual Feed Intake Holstein Dairy Cows. Metabolites 2021, 11, 868. [Google Scholar] [CrossRef]
- Pralle, R.S.; Li, W.; Murphy, B.N.; Holdorf, H.T.; White, H.M. Novel Facets of the Liver Transcriptome Are Associated with the Susceptibility and Resistance to Lipid-Related Metabolic Disorders in Periparturient Holstein Cows. Animals 2021, 11, 2558. [Google Scholar] [CrossRef] [PubMed]
- Arneson, A.; Haghani, A.; Thompson, M.J.; Pellegrini, M.; Kwon, S.B.; Vu, H.; Maciejewski, E.; Yao, M.; Li, C.Z.; Lu, A.T.; et al. A Mammalian Methylation Array for Profiling Methylation Levels at Conserved Sequences. Nat. Commun. 2022, 13, 783. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zoller, J.A.; Haghani, A.; Lu, A.T.; Raj, K.; Jasinska, A.J.; Mattison, J.A.; Salmon, A.B. DNA Methylation Age Analysis of Rapamycin in Common Marmosets. GeroScience 2021, 43, 2413–2425. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zoller, J.A.; Haghani, A.; Jasinska, A.J.; Raj, K.; Breeze, C.E.; Ernst, J.; Vaughan, K.L.; Mattison, J.A. Epigenetic Clock and Methylation Studies in the Rhesus Macaque. GeroScience 2021, 43, 2441–2453. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Haghani, A.; Peng, S.; Hales, E.N.; Zoller, J.A.; Raj, K.; Larison, B.; Robeck, T.R.; Petersen, J.L.; Bellone, R.R.; et al. DNA Methylation Aging and Transcriptomic Studies in Horses. Nat. Commun. 2022, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Haghani, A.; Macoretta, N.; Ablaeva, J.; Zoller, J.A.; Li, C.Z.; Zhang, J.; Takasugi, M.; Zhao, Y.; Rydkina, E.; et al. DNA Methylation Clocks Tick in Naked Mole Rats but Queens Age More Slowly than Nonbreeders. Nat. Aging 2022, 2, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Prado, N.A.; Brown, J.L.; Zoller, J.A.; Haghani, A.; Yao, M.; Bagryanova, L.R.; Campana, M.G.; Maldonado, J.E.; Raj, K.; Schmitt, D.; et al. Epigenetic Clock and Methylation Studies in Elephants. Aging Cell 2021, 20, e13414. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.; Szladovits, B.; Haghani, A.; Zoller, J.A.; Li, C.Z.; Black, P.; Maddox, D.; Robeck, T.R.; Horvath, S. Epigenetic Clock and Methylation Studies in Cats. GeroScience 2021, 43, 2363–2378. [Google Scholar] [CrossRef]
- Jasinska, A.J.; Haghani, A.; Zoller, J.A.; Li, C.Z.; Arneson, A.; Ernst, J.; Kavanagh, K.; Jorgensen, M.J.; Mattison, J.A.; Wojta, K.; et al. Epigenetic Clock and Methylation Studies in Vervet Monkeys. GeroScience 2022, 44, 699–717. [Google Scholar] [CrossRef]
- Lemaître, J.-F.; Rey, B.; Gaillard, J.-M.; Régis, C.; Gilot-Fromont, E.; Débias, F.; Duhayer, J.; Pardonnet, S.; Pellerin, M.; Haghani, A.; et al. DNA Methylation as a Tool to Explore Ageing in Wild Roe Deer Populations. Mol. Ecol. Resour. 2022, 22, 1002–1015. [Google Scholar] [CrossRef]
- Mozhui, K.; Lu, A.T.; Li, C.Z.; Haghani, A.; Sandoval-Sierra, J.V.; Wu, Y.; Williams, R.W.; Horvath, S. Genetic Loci and Metabolic States Associated with Murine Epigenetic Aging. eLife 2022, 11, e75244. [Google Scholar] [CrossRef]
- Connor, E.E.; Hutchison, J.L.; Van Tassell, C.P.; Cole, J.B. Defining the Optimal Period Length and Stage of Growth or Lactation to Estimate Residual Feed Intake in Dairy Cows. J. Dairy Sci. 2019, 102, 6131–6143. [Google Scholar] [CrossRef]
- Hardie, L.C.; VandeHaar, M.J.; Tempelman, R.J.; Weigel, K.A.; Armentano, L.E.; Wiggans, G.R.; Veerkamp, R.F.; de Haas, Y.; Coffey, M.P.; Connor, E.E.; et al. The Genetic and Biological Basis of Feed Efficiency in Mid-Lactation Holstein Dairy Cows. J. Dairy Sci. 2017, 100, 9061–9075. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinforma. Oxf. Engl. 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Triche, T.J., Jr.; Laird, P.W.; Shen, H. SeSAMe: Reducing Artifactual Detection of DNA Methylation by Infinium BeadChips in Genomic Deletions. Nucleic Acids Res. 2018, 46, e123. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Jorg, R. topGO: Enrichment Analysis for Gene Ontology Version 2.42.0 from Bioconductor. Available online: https://rdrr.io/bioc/topGO/ (accessed on 1 August 2023).
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.J.; Pairo-Castineira, E.; et al. A Multi-Tissue Atlas of Regulatory Variants in Cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Parsons, C.T.; Dafoe, J.M.; Wyffels, S.A.; DelCurto, T.; Boss, D.L. Influence of Residual Feed Intake and Cow Age on Dry Matter Intake Post-Weaning and Peak Lactation of Black Angus Cows. Animals 2021, 11, 1822. [Google Scholar] [CrossRef]
- Boj, S.F.; van Es, J.H.; Huch, M.; Li, V.S.W.; José, A.; Hatzis, P.; Mokry, M.; Haegebarth, A.; van den Born, M.; Chambon, P.; et al. Diabetes Risk Gene and Wnt Effector Tcf7l2/TCF4 Controls Hepatic Response to Perinatal and Adult Metabolic Demand. Cell 2012, 151, 1595–1607. [Google Scholar] [CrossRef]
- Wong, K.K.L.; Liu, T.-W.; Parker, J.M.; Sinclair, D.A.R.; Chen, Y.-Y.; Khoo, K.-H.; Vocadlo, D.J.; Verheyen, E.M. The Nutrient Sensor OGT Regulates Hipk Stability and Tumorigenic-like Activities in Drosophila. Proc. Natl. Acad. Sci. USA 2020, 117, 2004–2013. [Google Scholar] [CrossRef]
- Lagerlöf, O.; Slocomb, J.E.; Hong, I.; Aponte, Y.; Blackshaw, S.; Hart, G.W.; Huganir, R.L. The Nutrient Sensor OGT in PVN Neurons Regulates Feeding. Science 2016, 351, 1293–1296. [Google Scholar] [CrossRef]
- Morales, E.; Porta, M.; Vioque, J.; López, T.; Mendez, M.A.; Pumarega, J.; Malats, N.; Crous-Bou, M.; Ngo, J.; Rifà, J.; et al. Food and Nutrient Intakes and K-ras Mutations in Exocrine Pancreatic Cancer. J. Epidemiol. Community Health 2007, 61, 641–649. [Google Scholar] [CrossRef]
- El Asri, A.; Zarrouq, B.; El Kinany, K.; Bouguenouch, L.; Ouldim, K.; El Rhazi, K. Associations between Nutritional Factors and KRAS Mutations in Colorectal Cancer: A Systematic Review. BMC Cancer 2020, 20, 696. [Google Scholar] [CrossRef]
- Beunders, G.; Voorhoeve, E.; Golzio, C.; Pardo, L.M.; Rosenfeld, J.A.; Talkowski, M.E.; Simonic, I.; Lionel, A.C.; Vergult, S.; Pyatt, R.E.; et al. Exonic Deletions in AUTS2 Cause a Syndromic Form of Intellectual Disability and Suggest a Critical Role for the C Terminus. Am. J. Hum. Genet. 2013, 92, 210–220. [Google Scholar] [CrossRef]
- Beunders, G.; de Munnik, S.A.; Van der Aa, N.; Ceulemans, B.; Voorhoeve, E.; Groffen, A.J.; Nillesen, W.M.; Meijers-Heijboer, E.J.; Frank Kooy, R.; Yntema, H.G.; et al. Two Male Adults with Pathogenic AUTS2 Variants, Including a Two-Base Pair Deletion, Further Delineate the AUTS2 Syndrome. Eur. J. Hum. Genet. 2015, 23, 803–807. [Google Scholar] [CrossRef]
- Novotný, A.; Plavec, J.; Kocman, V. Structural Polymorphism Driven by a Register Shift in a CGAG-Rich Region Found in the Promoter of the Neurodevelopmental Regulator AUTS2 Gene. Nucleic Acids Res. 2023, 51, 2602–2613. [Google Scholar] [CrossRef] [PubMed]
- Graber, M.; Kohler, S.; Kaufmann, T.; Doherr, M.G.; Bruckmaier, R.M.; van Dorland, H.A. A Field Study on Characteristics and Diversity of Gene Expression in the Liver of Dairy Cows during the Transition Period. J. Dairy Sci. 2010, 93, 5200–5215. [Google Scholar] [CrossRef]
- Powell, I.C. Characterization of a No Acterization of a Novel Glycerol-3-Phosphate Dehy Ol-3-Phosphate Dehydrogenase Ogenase (GPD2) in the Alga Chlamydomonas reinhardtii. Master’s Thesis, University of Nebraska-Lincoln, Lincoln, NE, USA, 2021. [Google Scholar]
- Li, H.; Rai, M.; Buddika, K.; Sterrett, M.C.; Luhur, A.; Mahmoudzadeh, N.H.; Julick, C.R.; Pletcher, R.C.; Chawla, G.; Gosney, C.J.; et al. Lactate Dehydrogenase and Glycerol-3-Phosphate Dehydrogenase Cooperatively Regulate Growth and Carbohydrate Metabolism during Drosophila melanogaster Larval Development. Development 2019, 146, dev175315. [Google Scholar] [CrossRef] [PubMed]
- Kenny, D.A.; Fitzsimons, C.; Waters, S.M.; McGee, M. Invited Review: Improving Feed Efficiency of Beef Cattle—the Current State of the Art and Future Challenges. Animal 2018, 12, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The Molecular Hallmarks of Epigenetic Control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Fang, L.; Zhou, Y.; Santos, D.J.A.; Xiang, R.; Daetwyler, H.D.; Chamberlain, A.J.; Cole, J.B.; Li, C.; Yu, Y.; et al. Analyses of Inter-Individual Variations of Sperm DNA Methylation and Their Potential Implications in Cattle. BMC Genom. 2019, 20, 888. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.; Hu, Y.; Fang, L.; Gao, Y.; Xia, H.; Schroeder, S.G.; Rosen, B.D.; Connor, E.E.; Li, C.; et al. Comparative Whole Genome DNA Methylation Profiling across Cattle Tissues Reveals Global and Tissue-Specific Methylation Patterns. BMC Biol. 2020, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.; Talenti, A.; Fisch, A.; Hemmink, J.D.; Paxton, E.; Toye, P.; Santos, I.; Ferreira, B.R.; Connelley, T.K.; Morrison, L.J.; et al. Profiling the Immune Epigenome across Global Cattle Breeds. Genome Biol. 2023, 24, 127. [Google Scholar] [CrossRef] [PubMed]
- Capra, E.; Lazzari, B.; Milanesi, M.; Nogueira, G.P.; Garcia, J.F.; Utsunomiya, Y.T.; Ajmone-Marsan, P.; Stella, A. Comparison between Indicine and Taurine Cattle DNA Methylation Reveals Epigenetic Variation Associated to Differences in Morphological Adaptive Traits. Epigenetics 2023, 18, 2163363. [Google Scholar] [CrossRef] [PubMed]
- Elolimy, A.A.; Abdelmegeid, M.K.; McCann, J.C.; Shike, D.W.; Loor, J.J. Residual Feed Intake in Beef Cattle and Its Association with Carcass Traits, Ruminal Solid-Fraction Bacteria, and Epithelium Gene Expression. J. Anim. Sci. Biotechnol. 2018, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Khansefid, M.; Millen, C.A.; Chen, Y.; Pryce, J.E.; Chamberlain, A.J.; Vander Jagt, C.J.; Gondro, C.; Goddard, M.E. Gene Expression Analysis of Blood, Liver, and Muscle in Cattle Divergently Selected for High and Low Residual Feed Intake. J. Anim. Sci. 2017, 95, 4764–4775. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-Wide Association Studies for Common Human Diseases. Nat. Rev. Genet. 2011, 12, 529–541. [Google Scholar] [CrossRef]
- Devos, J.; Behrouzi, A.; Paradis, F.; Straathof, C.; Li, C.; Colazo, M.; Block, H.; Fitzsimmons, C. Genetic Potential for Residual Feed Intake and Diet Fed during Early- to Mid-Gestation Influences Post-Natal DNA Methylation of Imprinted Genes in Muscle and Liver Tissues in Beef Cattle. J. Anim. Sci. 2021, 99, skab140. [Google Scholar] [CrossRef]
- Andrés, S.; Madsen, O.; Montero, O.; Martín, A.; Giráldez, F.J. The Role of Feed Restriction on DNA Methylation, Feed Efficiency, Metabolome, Biochemical Profile, and Progesterone Patterns in the Female Filial Generation (F1) Obtained From Early Feed Restricted Ewes (F0). Front. Physiol. 2021, 12, 779054. [Google Scholar] [CrossRef]
- Urich, M.A.; Nery, J.R.; Lister, R.; Schmitz, R.J.; Ecker, J.R. MethylC-Seq Library Preparation for Base-Resolution Whole-Genome Bisulfite Sequencing. Nat. Protoc. 2015, 10, 475–483. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Boschiero, C.; Li, C.-J.; Connor, E.E.; Baldwin, R.L., VI; Liu, G.E. Unraveling the Genetic Basis of Feed Efficiency in Cattle through Integrated DNA Methylation and CattleGTEx Analysis. Genes 2023, 14, 2121. https://doi.org/10.3390/genes14122121
Hu Z, Boschiero C, Li C-J, Connor EE, Baldwin RL VI, Liu GE. Unraveling the Genetic Basis of Feed Efficiency in Cattle through Integrated DNA Methylation and CattleGTEx Analysis. Genes. 2023; 14(12):2121. https://doi.org/10.3390/genes14122121
Chicago/Turabian StyleHu, Zhenbin, Clarissa Boschiero, Cong-Jun Li, Erin E. Connor, Ransom L. Baldwin, VI, and George E. Liu. 2023. "Unraveling the Genetic Basis of Feed Efficiency in Cattle through Integrated DNA Methylation and CattleGTEx Analysis" Genes 14, no. 12: 2121. https://doi.org/10.3390/genes14122121
APA StyleHu, Z., Boschiero, C., Li, C.-J., Connor, E. E., Baldwin, R. L., VI, & Liu, G. E. (2023). Unraveling the Genetic Basis of Feed Efficiency in Cattle through Integrated DNA Methylation and CattleGTEx Analysis. Genes, 14(12), 2121. https://doi.org/10.3390/genes14122121