
Chloroplast Genome Comparison and Phylogenetic Analysis of the Commercial Variety Actinidia chinensis ‘Hongyang’

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Extraction of cpDNA, and Sequencing
2.2. CpDNA Assembly and Annotation
2.3. Analysis of the Cp Genome for Repetitive Sequences
2.4. Codon Preference Analysis
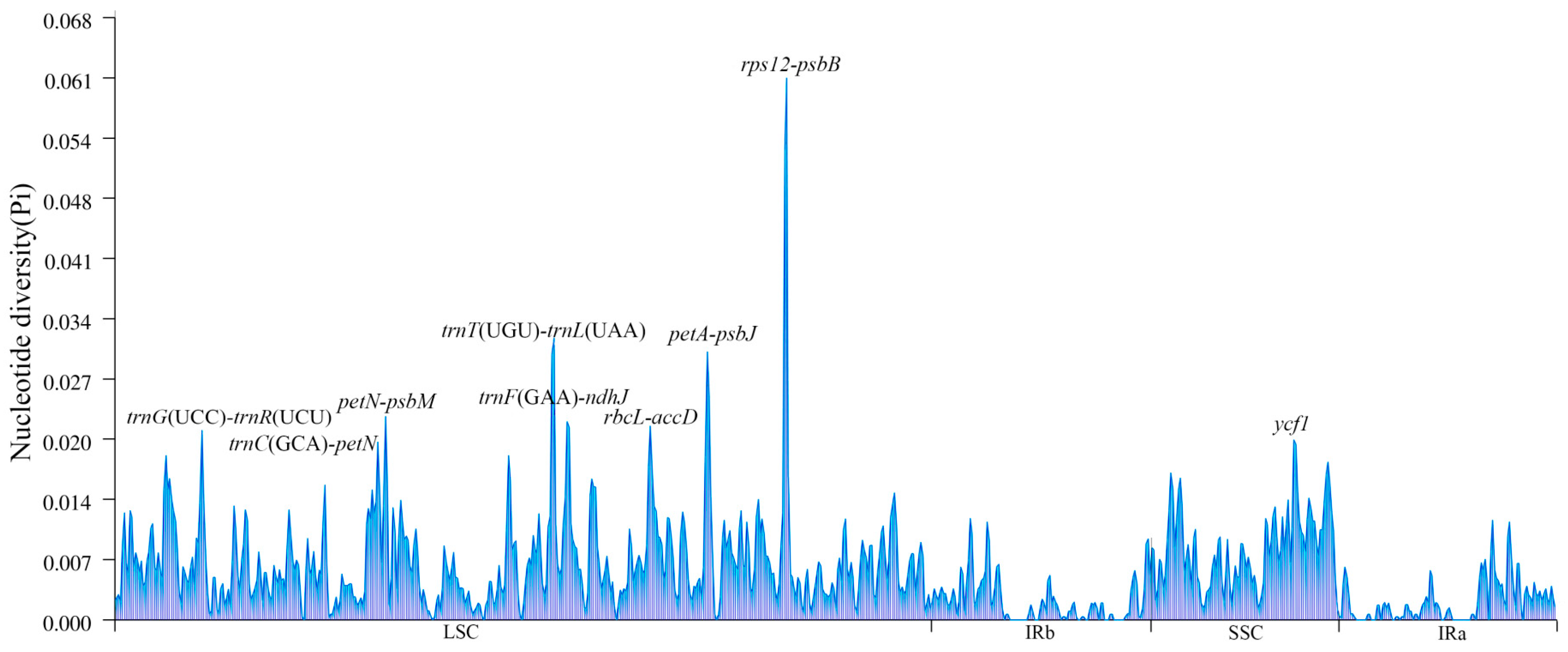
2.5. Sequence Variation Analysis
2.6. Phylogenetic Relationship Analysis
3. Results
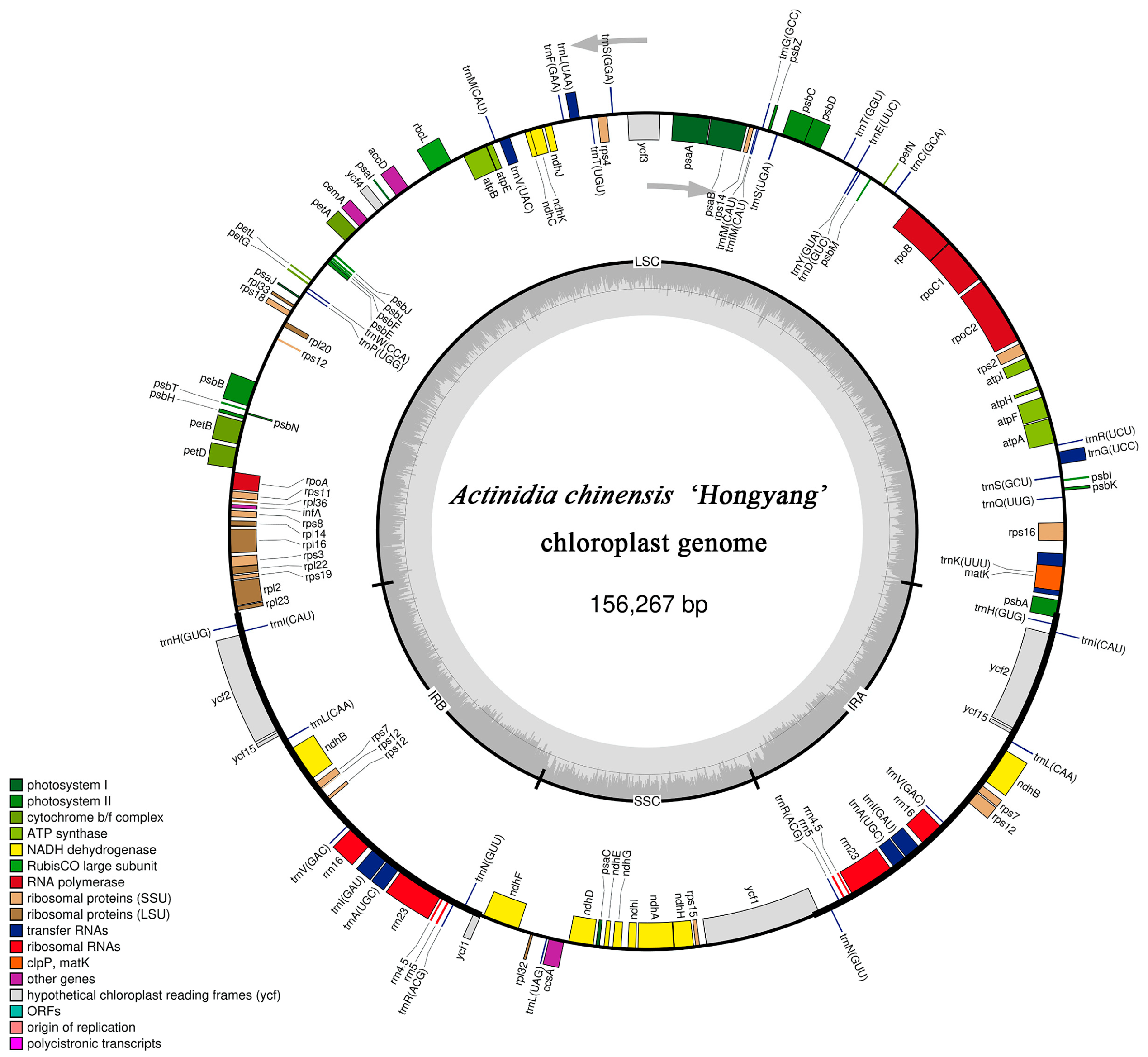
3.1. Chloroplast Genomic Structure of A. chinensis ’Hongyang’
3.2. Chloroplast Genome Composition of Actinidia
3.3. Codon Usage in the Chloroplast Genome of A. chinensis ’Hongyang’
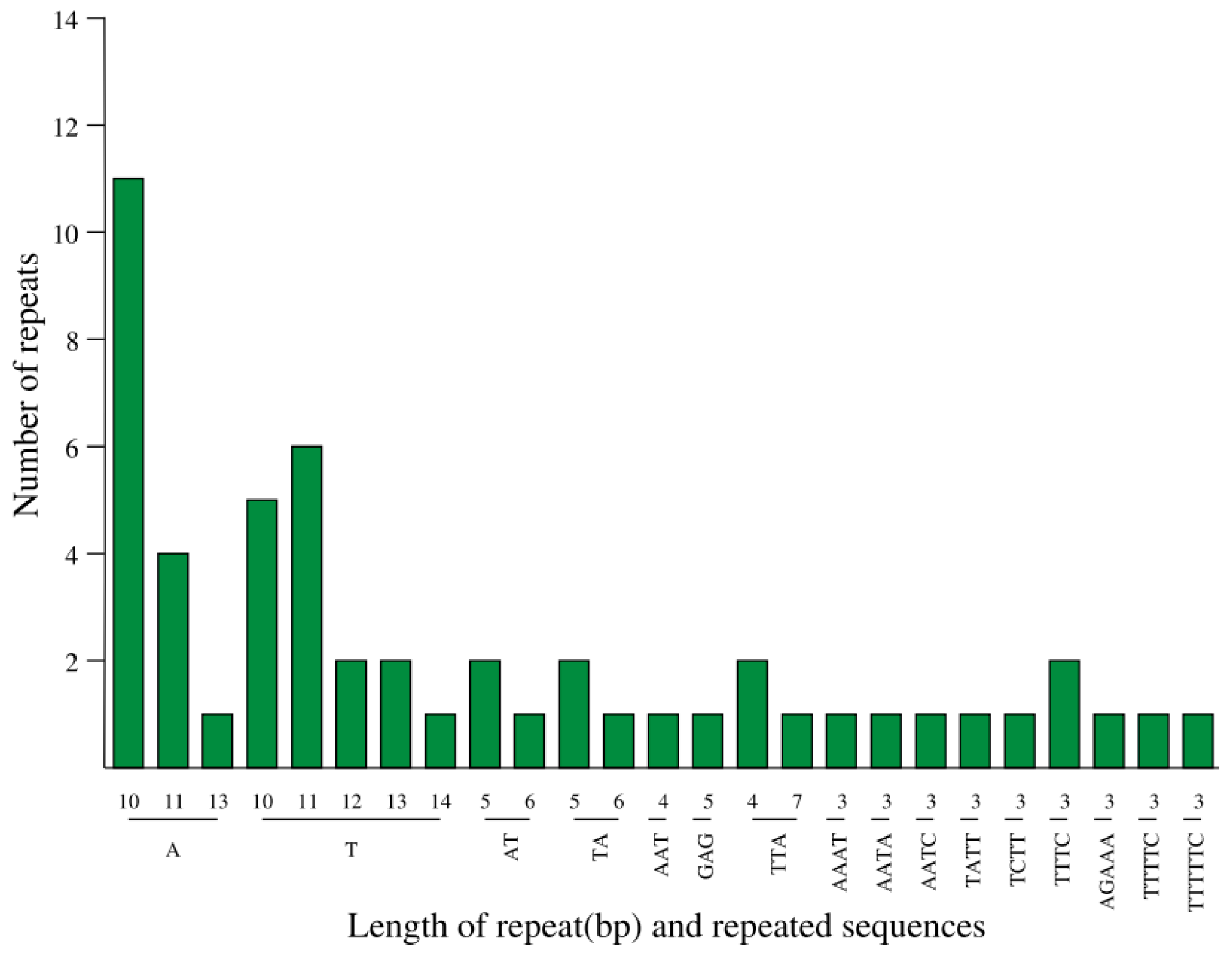
3.4. Detection of Repeated Sequences and SSRs in A. chinensis ‘Hongyang’ cpDNA
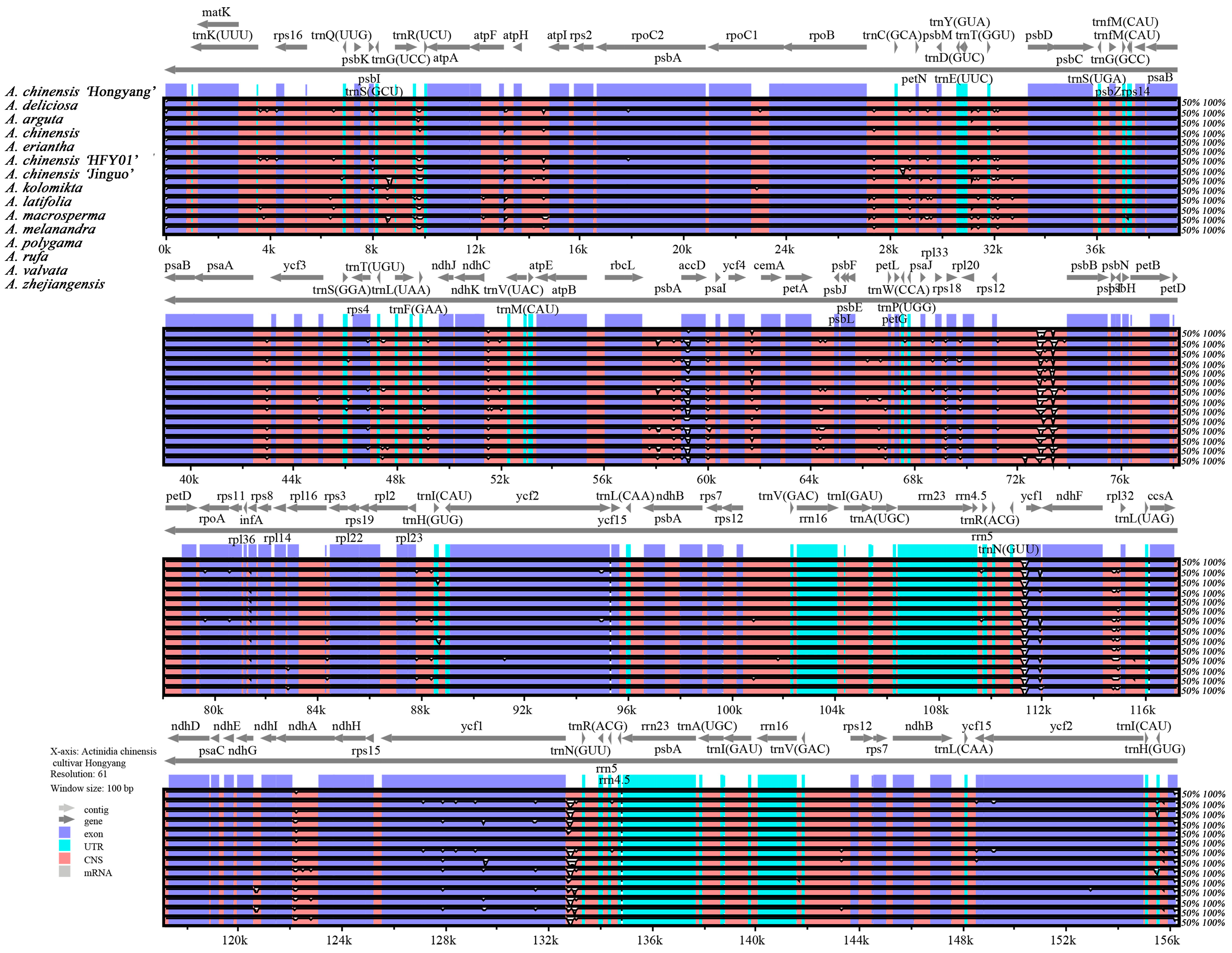
3.5. Comparative Analysis of Fifteen Species Chloroplast Genomic Structure
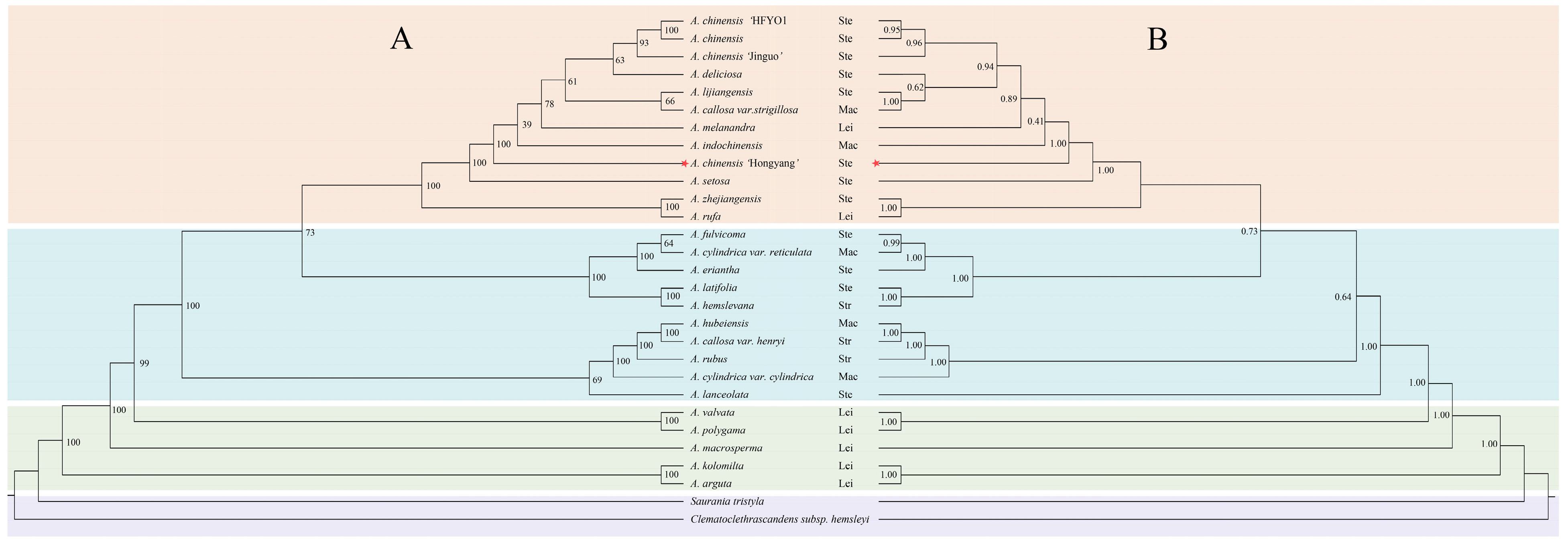
3.6. Phylogenetic Analysis of 29 Taxa of the Actinidiaceae Based on cpDNA Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, J.Y.; Pan, D.L.; Jia, Z.H.; Wang, T.; Wang, G.; Guo, Z.R. Chlorophyll, carotenoid and vitamin C metabilism regulation in Actinidia chinensis ‘Hongyang’ outer pericarp during fruit development. PLoS ONE 2018, 13, e0194835. [Google Scholar]
- He, J.L.; Wu, D.T.; Zhang, Q.; Chen, H.; Li, H.Y.; Han, Q.H.; Lai, X.Y.; Wang, H.; Wu, Y.X.; Yuan, J.G.; et al. Efficacy and mechanism of cinnamon essential oil on inhibition of colletotrichum acutatum isolated from ‘Hongyang’. Front. Microbiol. 2018, 9, 1288. [Google Scholar] [CrossRef]
- Hunter, D.C.; Greenwood, J.; Zhang, J.L.; Skinner, M.A. Antioxidant and ‘natural protective’ properties of Kiwifruit. Curr. Top. Med. Chem. 2011, 11, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, N.; Shirataki, Y.; Kawase, M.; Tani, S.; Sakagami, H.; Satoh, K.; Kurihara, T.; Nakashima, H.; Mucsi, I.; Varga, A.; et al. Cancer prevention and therapy with kiwifruit in Chinese folklore medicine: A study of kiwifruit extracts. J. Ethnopharmacol. 2002, 81, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.H.; Tang, P.; Li, Z.A.; Li, D.W.; Liu, Y.F.; Huang, H.W. The first complete chloroplast genome sequences in Actinidiaceae: Genome structure and comparative analysis. PLoS ONE 2015, 10, e0129347. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Chen, S.Y.; Zhang, X.Z. Chloroplast Genome Evolution in Actinidiaceae: clpP loss, heterogenous divergence and phylogenomic practice. PLoS ONE 2016, 11, e0162324. [Google Scholar] [CrossRef]
- Tang, P.; Shen, R.N.; He, R.W.; Yao, X.H. The complete chloroplast genome sequence of Actinidia eriantha. Mitochondrial DNA B 2019, 4, 2114–2115. [Google Scholar] [CrossRef]
- Lan, Y.; Cheng, L.; Huang, W.; Cao, Q.; Zhou, Z.; Luo, A.; Hu, G. The complete chloroplast genome sequence of Actinidia kolomikta from North China. Conserv. Genet. Resour. 2018, 10, 475–477. [Google Scholar] [CrossRef]
- Zhang, J.L.; Liu, H. The complete choloroplast genome sequence of Actinidia lanceolata. Mitochondrial DNA B 2019, 4, 1187–1188. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef]
- Daniell, H.; Wurdack, K.J.; Kanagaraj, A.; Lee, S.B.; Saski, C.; Jansen, R.K. The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II intron. Theor. Appl. Genet. 2008, 116, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Cui, L.; Depamphilis, C.W.; Moret, B.M.E.; Tang, J. Gene rearrangement analysis and ancestral order inference from chloroplast genomes with inverted repeat. BMC Genom. 2008, 9 (Suppl. 1), S25. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, J.; Cherney, B.; Merlin, E.; Williams, C.; Mets, L. Recombination within the inverted repeat sequences of the Chlamydomonas reinhardii chloroplast genome produces two orientation isomers. Curr. Genet. 1985, 9, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, J.; Cherney, B.W.; Williams, C. Sequence analysis of the junction of the large single copy region and the large inverted repeat in the petunia chloroplast genome. Curr. Genet. 1988, 14, 487–492. [Google Scholar] [CrossRef]
- Jian, H.Y.; Zhang, Y.H.; Yan, H.J.; Qiu, X.Q.; Wang, Q.G.; Li, S.B.; Zhang, S.D. The complete chloroplast genome of a key ancestor of modern roses, Rosa chinensis var. spontanea, and a comparison with congeneric species. Molecules 2018, 23, 389. [Google Scholar] [CrossRef]
- Saina, J.K.; Li, Z.Z.; Gichira, A.W.; Liao, Y.Y. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an important pantropical tree. Int. J. Mol. Sci. 2018, 19, 929. [Google Scholar] [CrossRef]
- Ping, T.; Qiang, X.; Ruinan, S.; Xiao, H.Y. Phylogenetic relationship in Actinidia (Actinidiaceae) based on four noncoding chloroplast DNA sequences. Plant. Syst. Evol. 2019, 305, 787–796. [Google Scholar]
- Ma, P.F.; Zhang, Y.X.; Zeng, C.X.; Guo, Z.H.; Li, D.Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef]
- Ruban, A.; Schmutzer, T.; Scholz, U.; Houben, A. How next-generation sequencing has aided our understanding of the sequence composition and origin of B chromosomes. Genes 2017, 8, 294. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-fast All-In-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with dogma. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. Organellargenomedraw (ogdraw): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Alzohairy, A.M. Bioedit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The codon adaptation index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Dong, W.P.; Xu, C.; Li, D.L.; Jin, X.B.; Li, R.L.; Lu, Q.; Suo, Z.L. Comparative analysis of the complete chloroplast genome sequences in psammophytic haloxylon species (Amaranthaceae). PeerJ 2016, 4, e2699. [Google Scholar] [CrossRef]
- Jiao, Y.; Jia, H.M.; Li, X.W.; Chai, M.L.; Jia, H.J.; Chen, Z.; Wang, G.Y.; Chai, C.Y.; Weg, E.V.D.; Gao, Z.S. Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genom. 2012, 13, 201. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Cato, S.A.; Richardson, T.E. Inter-and intraspecific polymorphism at chloroplast SSR loci and the inheritance of plastids in Pinus radiata D. Don. Theor. Appl. Genet. 1996, 93, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Somaratne, Y.; Guan, D.L.; Wang, W.Q.; Zhao, L.; Xu, S.Q. The complete chloroplast genomes of two Lespedeza species: Insights into codon usage bias, RNA editing sites, and phylogenetic relationships in Desmodieae (Fabaceae: Papilionoideae). Plants 2019, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Lee, J.W.; Baek, S.H.; Lee, M.W.; Kang, Y.J. The complete chloroplast genome sequence of Actinidia rufa (Actinidiaceae). Mitochondrial DNA B 2018, 3, 564–565. [Google Scholar] [CrossRef]
- Xiong, A.S.; Peng, R.H.; Zhuang, J.; Gao, F.; Zhu, B.; Fu, X.Y.; Xue, Y.; Jin, X.F.; Tian, Y.S.; Zhao, W.; et al. Gene duplication, transfer, and evolution in the chloroplast genome. Biotechnol. Adv. 2009, 27, 340–347. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Li, D.M.; Li, J.; Wang, D.R.; Xu, Y.C.; Zhu, G.F. Molecular evolution of chloroplast genomes in subfamily Zingiberoideae (Zingiberaceae). BMC Plant. Biol. 2021, 21, 558. [Google Scholar] [CrossRef]
- Lee, C.; Ruhlman, T.A.; Jansen, R.K. Unprecedented intraindividual structural heteroplasmy in Eleocharis (Cyperaceae, Poales) plastomes. Genome Biol. Evol. 2020, 12, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.Z.; Cai, X.D.; Gong, M.; Xia, M.Q.; Xing, H.T.; Dong, S.S.; Tian, S.M.; Li, J.L.; Lin, J.Y.; Liu, Y.Q.; et al. Complete chloroplast genomes provide insights into evolution and phylogeny of Zingiber (Zingiberaceae). BMC Genom. 2023, 24, 30. [Google Scholar]
- Peng, J.Y.; Zhang, X.S.; Zhang, D.G.; Wang, Y.; Deng, T.; Huang, X.H.; Kuang, T.H.; Zhou, Q. Newly reported chloroplast genome of Sinosenecio albonervius y. Liu & Q.E. Yang and comparative analyses with other Sinosenecio species. BMC Genom. 2022, 23, 639. [Google Scholar]
- Li, L.; Hu, Y.F.; He, M.; Zhang, B.; Wu, W.; Cai, P.; Huo, D.; Hong, Y.C. Comparative chloroplast genomes: Insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genom. 2021, 22, 138. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant. Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Abdullah; Mehmood, F.; Rahim, A.; Heidari, P.; Ahmed, I.; Poczai, P. Comparative plastome analysis of Blumea, with implications for genome evolution and phylogeny of Asteroideae. Ecol. Evol. 2021, 11, 7810–7826. [Google Scholar] [CrossRef]
- Liu, Y.F.; Li, D.W.; Zhang, Q.; Song, C.; Zhong, C.H.; Zhang, X.D.; Wang, Y.; Yao, X.H.; Wang, Z.P.; Zeng, S.H.; et al. Rapid radiations of both kiwifruit hybrid lineages and their parents shed light on a two-layer mode of species diversification. New. Phytologist. 2017, 215, 877–890. [Google Scholar] [CrossRef]
- Xiong, B.; Wang, T.; Huang, S.J.; Liao, L.; Wang, X.; Deng, H.H.; Zhang, M.F.; He, J.X.; Sun, G.C.; He, S.Y.; et al. Analysis of codon usage bias in xyloglucan endotransglycosylase (XET) genes. Int. J. Mol. Sci. 2023, 24, 6108. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Nie, X.J.; Lv, S.Z.; Zhang, Y.X.; Du, X.H.; Wang, L.; Biradar, S.S.; Tan, X.F.; Wan, F.H.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef]
- Dang, Y.Y.; Yang, Y.; Li, Q.; Lu, J.J.; Li, X.W.; Wang, Y.T. Complete chloroplast genome sequence of poisonous and medicinal plant Datura stramonium: Organizations and implications for genetic engineering. PLoS ONE 2014, 9, e110656. [Google Scholar]
- Chen, Q.; Wu, X.B.; Zhang, D.Q. Phylogenetic analysis of Fritillaria cirrhosa D. Don and its closely related species based on complete chloroplast genomes. PeerJ 2019, 7, e7480. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W.; Cowan, R.S.; Hollingsworth, P.M.; Berg, C.; Wilkinson, M.J. A proposal for a standardised protocol to barcode all land plants. Taxon 2007, 56, 295–299. [Google Scholar] [CrossRef]
- He, L.; Qian, J.; Li, X.W.; Sun, Z.Y.; Xu, X.L.; Chen, S.L. Complete chloroplast genome of medicinal plant Lonicera japonica: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 2017, 22, 249. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.L.; Zhang, H.B.; Yan, F.; Yan, C.X.; Wei, S.G.; Lai, J.H.; Wang, Y.P.; Zhang, B. Morphology and molecular identification of twelve commercial varieties of Kiwifruit. Molecules 2019, 24, 888. [Google Scholar] [CrossRef] [PubMed]
- Blazier, J.C.; Jansen, R.K.; Mower, J.P.; Govindu, M.; Zhang, J.; Weng, M.L.; LRuhlman, T.A. Variable presence of the inverted repeat and plastome stability in Erodium. Ann. Bot. 2016, 117, 1209–1220. [Google Scholar] [CrossRef]
- Li, J.L.; Tang, J.M.; Zeng, S.Y.; Han, F.; Yuan, J.; Yu, J. Comparative plastid genomics of four Pilea (Urticaceae) species: Insight into interspecifc plastid genome diversity in Pilea. BMC Plant Biol. 2021, 21, 25. [Google Scholar] [CrossRef]
- Weng, M.L.; Ruhlman, T.A.; Jansen, R.K. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New. Phytol. 2017, 214, 842–851. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, T.; Kanwal, N.; Zhao, Y.M.; Bai, G.Q.; Zhao, G.F. Completion of eight Gynostemma bl. (Cucurbitaceae) chloroplast genomes: Characterization, comparative analysis, and phylogenetic relationships. Front. Plant. Sci. 2017, 8, 1583. [Google Scholar] [CrossRef]
- Chat, J.; Jáuregui, B.; Petit, R.J.; Nadot, S. Reticulate evolution in kiwifruit (Actinidia, Actinidiaceae) identified by comparing their maternal and paternal phylogenies. Am. J. Bot. 2004, 91, 736–747. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | A (%) | C (%) | G (%) | T (U) (%) | AT (%) | GC (%) |
|---|---|---|---|---|---|---|
| LSC | 31.53 | 18.16 | 17.34 | 32.97 | 64.50 | 35.50 |
| SSC | 33.83 | 16.67 | 14.48 | 35.02 | 68.85 | 31.15 |
| IRs | 28.78 | 20.57 | 22.31 | 28.33 | 57.11 | 42.89 |
| Total | 30.92 | 18.98 | 18.23 | 31.88 | 62.80 | 37.20 |
| Species | GenBank Number | Genome Size (bp) | LSC (bp) | SSC (bp) | IR (bp) | Genes | PCGs | tRNAs | rRNAs | GC Content (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| A. chinensis | 156,216 | 88,316 | 20,332 | 23,784 | 132 | 86 | 38 | 8 | 37.20 | |
| A. arguta | 157,611 | 88,532 | 20,463 | 24,308 | 133 | 84 | 41 | 8 | 37.10 | |
| A. deliciosa | 156,741 | 88,267 | 20,332 | 24,071 | 130 | 83 | 39 | 8 | 37.20 | |
| A. eriantha | 156,964 | 88,639 | 20,541 | 23,892 | 131 | 84 | 39 | 8 | 37.20 | |
| A. chinensis ‘HFY01’ | 156,322 | 88,085 | 20,331 | 23,953 | 131 | 84 | 39 | 8 | 37.20 | |
| A. chinensis ’Jinguo’ | 156,743 | 88,265 | 20,332 | 24,073 | 132 | 85 | 39 | 8 | 37.20 | |
| A. kolomikta | 157,425 | 88,498 | 20,475 | 24,226 | 131 | 84 | 39 | 8 | 37.20 | |
| A. latifolia | 156,873 | 88,666 | 21,453 | 23,377 | 132 | 83 | 41 | 8 | 37.20 | |
| A. macrosperma | 156,231 | 88,214 | 20,577 | 23,720 | 132 | 85 | 39 | 8 | 37.20 | |
| A. melanandra | 156,124 | 88,006 | 20,332 | 23,893 | 131 | 84 | 39 | 8 | 37.20 | |
| A. polygama | 156,583 | 88,568 | 20,397 | 23,809 | 131 | 83 | 40 | 8 | 37.20 | |
| A. rufa | 156,543 | 88,436 | 20,307 | 23,900 | 131 | 84 | 39 | 8 | 37.20 | |
| A. valvata | 156,660 | 88,397 | 20,447 | 23,908 | 134 | 85 | 41 | 8 | 37.20 | |
| A. zhejiangensis | 156,717 | 86,483 | 22,574 | 23,840 | 128 | 81 | 39 | 8 | 37.20 | |
| A. chinensis ‘Hongyang’ | 156,267 | 87,866 | 20,335 | 24,033 | 132 | 85 | 39 | 8 | 37.20 |
| Category | Gene Group | Gene Name | Number |
|---|---|---|---|
| Photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ | 5 |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | 15 | |
| Subunits of NADH dehydrogenase | ndhA *, ndhB *(2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 12 | |
| Subunits of the cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | 6 | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | 6 | |
| Large subunit of rubisco | rbcL | 1 | |
| Self-replication | Proteins of large ribosomal subunits | rpl14, rpl16 *, rpl2 *, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 | 9 |
| Proteins of the small ribosomal subunit | rps11, rps12 **(2), rps14, rps15, rps16 *, rps18, rps19, rps2, rps3, rps4, rps7 (2), rps8 | 14 | |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | 4 | |
| Ribosomal RNAs | rrn16 (2), rrn23 (2), rrn4.5 (2), rrn5 (2) | 8 | |
| Transfer RNAs | trnA(UGC) *(2), trnC(GCA), trnD(GUC), trnE(UUC), trnF(GAA), trnG(GCC), trnG(UCC) *, trnH(GUG) (2), trnI(CAU) (2), trnI(GAU) *(2), trnK(UUU) *, trnL(CAA) (2), trnL(UAA) *, trnL(UAG), trnM(CAU), trnN(GUU) (2), trnP(UGG), trnQ(UUG), trnR(ACG) (2), trnR(UCU), trnS(GCU), trnS(GGA), trnS(UGA), trnT(GGU), trnT(UGU), trnV(GAC) (2), trnV(UAC) *, trnW(CCA), trnY(GUA), trnfM(CAU) (2) | 39 | |
| Other genes | Maturase | matK | 1 |
| Envelope membrane protein | cemA | 1 | |
| Acetyl-CoA carboxylase | accD | 1 | |
| c-type cytochrome synthesis gene | ccsA | 1 | |
| Translation initiation factor | infA | 1 | |
| Genes of unknown function | Conserved hypothetical cp open reading framework | ycf1 (2), ycf15 (2), ycf2 (2), ycf3 **, ycf4 | 8 |
| Total | 132 |
| Genes | Local | Exon1 | Intron1 | Exon2 | Intron2 | Exon3 |
|---|---|---|---|---|---|---|
| rps12 | IRa + LSC | 114 | 536 | 232 | 26 | |
| trnK(UUU) | LSC | 37 | 2489 | 39 | ||
| rps16 | LSC | 38 | 875 | 226 | ||
| trnG(UCC) | LSC | 23 | 689 | 48 | ||
| atpF | LSC | 145 | 733 | 410 | ||
| rpoC1 | LSC | 453 | 748 | 1605 | ||
| ycf3 | LSC | 126 | 710 | 228 | 743 | 153 |
| trnL(UAA) | LSC | 37 | 504 | 50 | ||
| trnV(UAC) | LSC | 39 | 595 | 37 | ||
| rps12-2 | IRb + LSC | 114 | 536 | 232 | 26 | |
| petB | LSC | 6 | 753 | 645 | ||
| petD | LSC | 8 | 777 | 475 | ||
| rpl16 | LSC | 9 | 1055 | 399 | ||
| rpl2 | LSC | 405 | 664 | 441 | ||
| ndhB | IRa | 777 | 679 | 756 | ||
| trnI(GAU) | IRa | 42 | 942 | 35 | ||
| trnA(UGC) | IRa | 38 | 798 | 35 | ||
| ndhA | SSC | 553 | 1085 | 539 | ||
| trnI(GAU)-2 | IRb | 42 | 942 | 35 | ||
| ndhB-2 | IRb | 777 | 679 | 756 | ||
| trnA(UGC)-2 | IRb | 38 | 798 | 35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Liu, X.; Sun, C.; Li, H.-L.; Li, Z.-X.; Guo, Y.; Fu, X.-Q.; Liao, Q.-H.; Zhang, W.-L.; Liu, Y.-Q. Chloroplast Genome Comparison and Phylogenetic Analysis of the Commercial Variety Actinidia chinensis ‘Hongyang’. Genes 2023, 14, 2136. https://doi.org/10.3390/genes14122136
Liu H, Liu X, Sun C, Li H-L, Li Z-X, Guo Y, Fu X-Q, Liao Q-H, Zhang W-L, Liu Y-Q. Chloroplast Genome Comparison and Phylogenetic Analysis of the Commercial Variety Actinidia chinensis ‘Hongyang’. Genes. 2023; 14(12):2136. https://doi.org/10.3390/genes14122136
Chicago/Turabian StyleLiu, Han, Xia Liu, Chong Sun, Hong-Lei Li, Zhe-Xin Li, Yuan Guo, Xue-Qian Fu, Qin-Hong Liao, Wen-Lin Zhang, and Yi-Qing Liu. 2023. "Chloroplast Genome Comparison and Phylogenetic Analysis of the Commercial Variety Actinidia chinensis ‘Hongyang’" Genes 14, no. 12: 2136. https://doi.org/10.3390/genes14122136